STIVARGA 40 mg 84 film kaplı tablet Farmakolojik Özellikler
{ Regorafenib }
5. FARMAKOLOJİK ÖZELLİKLER
5.1. Farmakodinamik özellikler
Farmakoterapötik grup: Diğer antineoplastik ajanlar, protein kinaz inhibitörleri
ATC kodu: L01EX05
Etki mekanizması ve farmakodinamik etkiler:
Regorafenib tümör anjiyogenezinde (VEGFR1, -2, -3, TIE2), onkogenezde (KIT, RET, RAF- 1, BRAF, BRAFV600E), metastazda (VEGFR3, PDGFR, FGFR) ve tümör immünitesinde (CSF1R) yer alan kinazlar dahil olmak üzere çoklu protein kinazları güçlü bir şekilde engelleyen bir oral tümör deaktivasyon ilacıdır. Regorafenib özellikle gastrointestinal stromal tümörlerde majör bir onkojenik etken olan mutasyona uğramış KIT'i inhibe eder ve
böylelikle tümör hücresi proliferasyonunu bloke eder. Klinik öncesi çalışmalarda, regorafenib
kolorektal, gastrointestinal stromal ve hepatoselüler tümör modelleri dahil olmak üzere
birçok tümör modelinde muhtemel antianjiyogenik ve antiproliferatif etkileri aracılığı ile güçlü anti-tümör aktivite göstermiştir. Ek olarak, regorafenib tümör ile ilişkili makrofajların seviyelerini düşürmüş ve in vivo anti-metastatik etki göstermiştir. Majör insan metabolitleri (M-2 ve M-5) in vitro ve in vivo modellerde regorafenib ile karşılaştırıldığında benzer etkililik sergilemişlerdir.
Klinik etkililik ve güvenlilik:
Metastatik kolorektal kanser (mKRK)
STIVARGA'nın klinik etkililiği ve güvenliliği, standart tedavi ile başarısızlık sonrasında progresyon göstermiş daha önce yoğun tedavi görmüş mKRK'li hastalar ile yapılan uluslararası, çok merkezli, randomize, çift kör, plasebo kontrollü bir faz III çalışmasında (CORRECT) değerlendirilmiştir.
Bu çalışmanın birincil etkililik sonlanım noktası genel sağkalımdır (OS). İkincil sonlanım noktaları progresyonsuz sağkalım (PFS), objektif tümör yanıt oranı ve hastalık kontrol oranıdır.
Toplamda, 760 hasta 2:1 oranında 3 hafta boyunca günde bir kez oral yolla uygulanan 160 mg regorafenib (her biri 40 mg regorafenib içeren 4 adet STIVARGA tablet) ile birlikte En İyi Destek Tedavi (EİDT) (N=505) ya da denk plasebo (N=255) ile birlikte EİDT uygulanmasını takiben 1 hafta tedavisiz periyoda randomize edilmiştir. Kullanılan ortalama günlük regorafenib dozu 147 mg'dır.
Hastalar, hastalık progresyonu veya kabul edilemeyen toksisite ortaya çıkana kadar tedaviye devam etmiştir. 432 ölüm meydana geldiğinde etkililik için önceden planlanmış bir interim analiz gerçekleştirilmiştir.
OS için planlanmış bu interim analiz, önceden belirlenmiş etkililik sınırını geçtikten sonra, yani plasebo ile birlikte uygulanan EİDT ile karşılaştırıldığında STIVARGA ile birlikte EİDT uygulanmasının sağkalımda uzama sağladığının kanıtı gösterildiğinde çalışmanın körlüğü kaldırılmıştır.
Randomize edilen 760 hastanın medyan yaşı 61 olup, %61'i erkek, %78'i beyaz ırktandır ve hastaların tamamının başlangıç ECOG (Eastern Cooperative Oncology Group) performans skoru (PS) 0 veya 1'dir. Hastaların %11,4'ünde STIVARGA tedavisi sırasında PS en az 2 bildirilmiştir.
Doz modifikasyonu ve doz azaltımı oranlarının yanı sıra medyan tedavi süresi ve günlük doz, plasebo alıp PS'nin en az 2 olarak bildirildiği hastalarda (%8,3) gözlemlenenle benzerdir. PS'nin en az 2 olduğu hastaların çoğunda tedavi hastalığın progresyonu nedeniyle durdurulmuştur. PS2 hastalar ve başlangıçtaki dehidratasyonu ≥1 olan hastalar, pivot çalışmanın dışında tutulmuştur. Birincil hastalık bölgesi kolon (%65), rektum (%29) veya her ikisidir (%6). Çalışmanın başlangıcında hastaların %57'sinde KRAS mutasyonu bildirilmiştir.
Hastaların çoğu (%52) metastatik hastalığın tedavisi için daha önce 3 veya daha az basamak tedavi almıştır. Bu tedaviler, floropirimidin içeren kemoterapi, anti-VEGF tedavisi, hasta KRAS doğal tip ise, anti-EGFR tedavisini içermiştir.
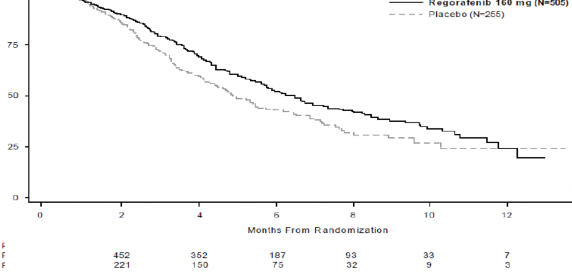
STIVARGA'nın EİDT'ye eklenmesi, plasebonun EİDT'ye eklenmesine kıyasla anlamlı olarak daha uzun sağkalım lasonuçlanmıştır; p değeri gruplanmış log-sıra testine göre
anlamlı olarak daha uzundur (tehlike oranı: 0,494, p<0,000001, bkz., Tablo 5 ve Şekil 2). Yanıt oranı (tam yanıt veya kısmi yanıt), STIVARGA ve plasebo ile tedavi edilen hastalarda sırasıyla (p= 0,188432, tek yönlü) %1 ve %0,4 olmuştur. Hastalık kontrol oranı (tam yanıt veya kısmi yanıt ya da stabil hastalık), STIVARGA ile tedavi edilen hastalarda anlamlı düzeyde daha yüksek olmuştur (%41,0 karşısında %14,9, p<0,000001, tek yönlü).
Tablo 5: CORRECT çalışmasından elde edilen etkililik sonuçları
Etkililik parametresi | Tehlike Oranı* (%95 GA) | P-değeri (tek taraflı) | Medyan (%95 GA) | |
STIVARGA + EİDT (N=505) | Plasebo + EİDT (N=255) | |||
Genel Sağkalım | 0,774 (0,636, 0,942) | 0,005178 | 6,4 ay (5,9, 7,3) | 5,0 ay (4,4, 5,8) |
Progresyonsuz Sağkalım** | 0,494 (0,419, 0,582) | <0,000001 | 1,9 ay (1,9, 2,1) | 1,7 ay (1,7, 1,7) |
* Tehlike oranı <1 STIVARGA lehine.
** Araştırmacının tümör yanıtını değerlendirmesine dayanır.
Randomizasyondan sonraki aylar
Şekil 1: Genel Sağkalım için Kaplan-Meier eğrileri
Genel sağkalım ve progresyonsuz sağkalım için yaş (<65; ≥65), cinsiyet, ECOG PS, primer hastalık yeri, metastatik hastalığın ilk tanısına kadar geçen süre, önceki antikanser tedavisi, metastatik hastalık için önceki tedavi basamakları ve KRAS mutasyon durumuna göre alt grup analizi, tedavi etkisinin plasebo rejimine kıyasla regorafenib rejimi lehine olduğunu göstermiştir.
Önceki KRAS mutasyon durumuna göre alt grup analizinin sonuçları, KRAS doğal tip tümörlü hastalarda GS açısından plaseboya kıyasla regorafenib lehine tedavi etkisi olduğunu göstermektedir. KRAS mutant tümörlü hastalarda ise sayıca daha az etki bildirilmiştir. PFS açısından KRAS mutasyon tipinden bağımsız olarak regorafenib lehine tedavi etkisi gözlemlenmiştir. Genel sağkalım tehlike oranı (%95 GA), KRAS doğal tip tümörlü hastalar için 0,653 (0,476 ila 0,895) şeklindeyken KRAS mutant tümörlü hastalarda 0,867 (0,670 ila 1,123) bulunmuştur. Tedavi etkisinde heterojenite kanıtı görülmemiştir (anlamlı olmayan
etkileşim testi). Progresyonsuz sağkalım tehlike oranı (%95 GA), KRAS doğal tip tümörlü
hastalar için 0,475 (0,362 ila 0,623) şeklindeyken KRAS mutant tümörlü hastalarda 0,525
(0,425 ila 0,649) bulunmuştur
İkinci bir faz III, uluslararası, çok merkezli, randomize, çift kör, plasebo kontrollü çalışmada (CONCUR) STIVARGA'nın etkililik ve güvenliliği önceden tedavi almış metastatik kolorektal kanseri olan ve floropirimidin bazlı kemoterapi başarısızlığından sonra progresyon görülen 204 Asyalı hastada (>%90 Doğu Asyalı) değerlendirilmiştir. CONCUR çalışmasındaki hastaların sadece %59,5'u öncesinde VEGF veya EGFR hedefli tedaviler ile tedavi edilmiştir.
Primer etkililik sonlanım noktası olarak OS değerlendirilmiştir. EİDT'ye STIVARGA eklenmesi plasebo artı EİDT'ye kıyasla anlamlı derecede daha uzun sağkalımla sonuçlanmış, tehlike oranının 0,550 (p = 0,000159 gruplanmış log-sıra testi) ve medyan GS'nin 8,8 aya kıyasla 6,3 ay olduğu [%95 GA 0,395, 0,765] belirlenmiştir.
STIVARGA artı EİDT alan hastalarda PFS'nin de anlamlı derecede daha uzun olduğu belirlenmiştir, medyan PFS STIVARGA ile 3,2 ayken plasebo ile 1,7 aydır. (tehlike oranı: 0,311, p<0,000001). CONCUR çalışmasında STIVARGA artı EİDT'nin güvenlilik profilinin CORRECT çalışmasında gözlenen güvenlilik profiliyle tutarlı olduğu görülmüştür.
Gastrointestinal stromal tümörler (GİST)
STIVARGA'nın klinik etkililiği ve güvenliliği, öncesinde 2 tirozin kinaz inhibitörü (imatinib ve sunitinib) ile tedavi görmüş GİST'li hastalar ile yapılan uluslararası, çok merkezli, randomize, çift kör, plasebo kontrollü bir faz III çalışmasında değerlendirilmiştir.
Progresyonsuz sağkalım (PFS) birincil etkililik sonlanım noktası analizi, 144 PFS vakasından sonra gerçekleştirilmiştir (merkezi körlenmiş değerlendirme). Progresyona kadar geçen süre (TTP) ve genel sağkalımdan (OS) oluşan ikincil sonlanım noktaları da değerlendirilmiştir (interim analiz).
Toplamda 199 GİST hastası, 2:1 oranında 3 hafta boyunca ya günde bir kez oral yolla uygulanan 160 mg regorafenib ile birlikte EİDT (n=133) ya da denk plasebo ile birlikte EİDT (n=66) uygulanmasını takiben 1 hafta tedavisiz periyoda randomize edilmiştir. Kullanılan ortalama günlük regorafenib dozu 140 mg'dır.
Hastalar, hastalık progresyonu veya kabul edilemeyen toksisite ortaya çıkana kadar tedaviye devam etmiştir. Plasebo kullanan ve hastalığında progresyon görülen hastalara açık etiketli regorafenib tedavisi (çapraz geçiş seçeneği) olanağı sunulmuştur. Regorafenib kullanan ve hastalığında progresyon görülen ve araştırmacının kanaatine göre regorafenib ile tedavinin klinik fayda sağlamakta olduğu hastalara açık etiketli regorafenibe devam etme imkanı verilmiştir.
Randomize edilen 199 hastanın ortalama yaşı 58 olup %64'ü erkek, %68'i beyaz ırktandır ve hastaların tamamının başlangıç ECOG Performans Skoru 0 veya 1'dir. En yakın progresyon veya relapstan bu yana geçen genel medyan süre 6 hafta olarak belirlenmiştir.
EİDT ile regorafenib kullanılması, plasebo ile birlikte uygulanan EİDT'ye kıyasla anlamlı derecede daha uzun PFS ile sonuçlanmış olup, tehlike oranı 0,268 [%95 GA 0,185, 0,388] ve medyan PFS 0,9 aya karşın 4,8 aydır (p<0,000001). Hastalık progresyonu veya ölüm için
bağıl risk regorafenib ile tedaviedilenhastalarda, plasebo ile tedavi edilen hastalar ile
yaş, cinsiyet, coğrafik bölge, önceki tedavi basamakları ve ECOG performans skorundan bağımsız olarak tutarlı olmuştur.
TTP, EİDT ile birlikte regorafenib kullanan hastalarda, EİDT ile birlikte plasebo uygulanan hastalara kıyasla anlamlı derecede daha uzun olmuş olup, tehlike oranı 0,248 [%95 GA 0,170, 0,364] ve medyan TTP 0,9 aya karşın 5,4 aydır (p<0,000001) (bkz., Tablo 6).
Başlangıçta plasebo koluna randomize edilmiş hastaların %85'inde progresyon sonrası çapraz geçiş gerçekleşmiş olmakla birlikte, OS analizinin tehlike oranı, pozitif tedavi etkisi yönünde eğilim göstermiştir (tehlike oranı=0,772 [%95 GA, 0,423, 1,408]; p=0,199; iki kolda da medyan OS'ye ulaşılamamıştır) (bkz., Tablo 6, Şekil 4).
Tablo 6 : GRID çalışmasından elde edilen etkililik sonuçları
Etkililik | Tehlike Oranı* | P-değeri | Medyan (%95 GA) | |
parametresi | (%95 GA) | (tek taraflı) | STIVARGA + EİDT (N=133) | Plasebo + EİDT (N=66) |
Progresyonsuz Sağkalım | 0,268 (0,185, 0,388) | <0,000001 | 4,8 ay (4,0, 5,7) | 0,9 ay (0,9, 1,1) |
Progresyona Kadar Geçen Süre | 0,248 (0,170, 0,364) | <0,000001 | 5,4 ay (4,1, 5,7) | 0,9 ay (0,9, 1,1) |
Genel Sağkalım | 0,772 (0,423, 1,408) | 0,199 | Ulaşılamamıştır | Ulaşılamamıştır |
GA: Güven aralığı
* Tehlike oranı <1 STIVARGA lehine.
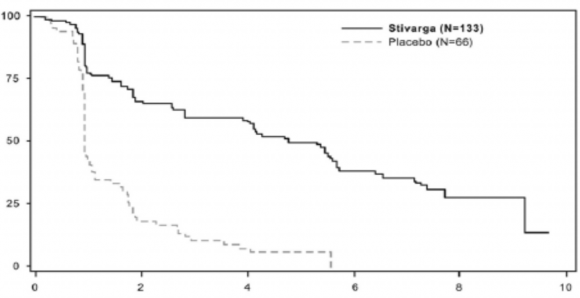
Şekil 2: Progresyonsuz Sağkalım için Kaplan-Meier eğrileri
Progresyonsuz Sağkalım Olasılığı (%)
Randomizasyondan sonraki aylar
![]()
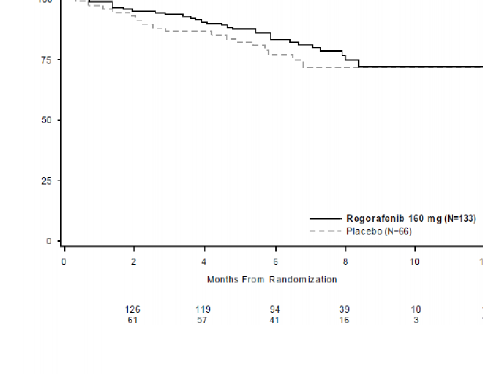
Genel Sağkalım Olasılığı (%)
Şekil 3: Genel sağkalım için Kaplan-Meier eğrileri
Ek olarak, EİDT ile plasebo uygulanan 56 hasta, hastalık progresyonunu takiben çapraz geçişten sonra açık etiketli STIVARGA kullanmıştır ve EİDT ile STIVARGA uygulanan toplam 41 hasta, hastalık progresyonunu takiben STIVARGA tedavisine devam etmiştir. Medyan ikincil PFS (araştırmacı değerlendirmelerine göre ölçüldüğünde) sırasıyla 5,0 ve 4,5 aydır.
Hepatoselüler karsinom (HSK)
STIVARGA'nın klinik etkililik ve güvenliliği, daha önce sorafenib tedavisi almış hepatoselüler karsinomlu hastalarla yürütülen uluslararası, çok merkezli, randomize, çift kör, plasebo kontrollü bir faz III çalışmada (RESORCE) değerlendirilmiştir.
Primer etkililik sonlanım noktası Genel Sağkalım (GS) olmuştur. Sekonder sonlanım noktaları Progresyonsuz Sağkalım (PFS), Progresyona Kadar Geçen Süre (TTP), Objektif Tümör Yanıt Oranı (ORR) ve Hastalık Kontrol Oranıdır (DCR).
Demografik özellikler ve başlangıç özellikleri STIVARGA ve plasebo tedavisi alan gruplarda
benzerdir ve randomize edilen 573 hasta için aşağıdaki tabloda gösterilmiştir:
Medyan yaş: 63 yaş
5.2. Farmakokinetik özellikler
Genel ÖzelliklerEmilim:
Her biri 40 mg içeren 4 tablet olarak verilen 160 mg'lık tek bir oral regorafenib dozundan yaklaşık 3 ila 4 saat sonra, regorafenib yaklaşık 2,5 mg/L'lik ortalama doruk plazma düzeyine ulaşır. Tabletlerin ortalama bağıl biyoyararlanımı oral çözeltiye kıyasla %69-83'tür. Regorafenib ve farmakolojik olarak aktif majör metabolitlerinin M-2 (N-oksit) ve M-5 (N- oksit ve N-desmetil) konsantrasyonları, ilacın az yağlı (hafif) bir kahvaltının ardından verilmesi durumunda yağdan zengin bir kahvaltı sonrasında ya da açlık koşullarında verilmesine kıyasla en yüksektir. Regorafenibe maruziyet açlık koşullarına kıyasla, ilacın yağdan zengin bir kahvaltı ile verilmesi durumunda %48, az yağlı bir kahvaltı ile verilmesi durumunda ise %36 artmıştır. M-2 ve M-5 metabolitlerine maruziyet, regorafenibin az yağlı bir kahvaltı ile verilmesi durumunda açlık durumuna kıyasla daha yüksek, yağdan zengin bir yemek ile verilmesi durumunda ise açlık koşullarında verilmesine kıyasla daha düşük olmuştur.
Dağılım:
Regorafenibin yanı sıra dolaşımdaki majör metabolitler için plazma konsantrasyon-zaman profilleri, 24 saatlik doz uygulama aralığında enterohepatik dolaşıma bağlı olarak çok sayıda pik göstermiştir.
Regorafenibin insan plazma proteinlerine in vitro protein bağlanması yüksektir (%99,5). Biyotransformasyon:
Regorafenib esas olarak karaciğerde metabolize olur, CYP3A4'ün aracılık ettiği oksidatif metabolizmaya girerken, aynı zamanda UGT1A9 aracılığıyla glukuronidasyona da uğrar. Plazmada iki majör ve altı minör regorafenib metaboliti tanımlanmıştır. Regorafenibin plazmada dolaşan esas metabolitleri, farmakolojik olarak aktif olan ve kararlı durumda regorafenib ile benzer konsantrasyonlara sahip M-2 (N-oksit) ve M-5 (N-oksit ve N- desmetil)'tir.
M-2 ve M-5'in in vitro proteine bağlanmaları regorafenibe göre daha yüksektir (sırasıyla
%99,8 ve %99,95).
Metabolitleri, gastrointestinal sistemde mikrobiyal flora tarafından indirgenebilir veya hidrolize olabilir ve bu da konjuge olmayan ilaç ve metabolitlerin geri emilmelerini sağlayabilir (enterohepatik dolaşım).
Eliminasyon:
Oral uygulamayı takiben, plazmadaki regorafenib ve metabolit M-2 için ortalama eliminasyon yarılanma ömrü farklı çalışmalarda 20 ila 30 saat arasında değişmektedir. Metabolit M-5 için ortalama eliminasyon yarılanma ömrü yaklaşık 60 saattir (40 ila 100 saat arasında).
Radyoaktif dozun yaklaşık %90'ı uygulamayı takiben 12 gün içinde geri kazanılmış ve dozun yaklaşık %71'i feçes ile (%47 ana bileşen, %24 metabolit olarak) ve dozun yaklaşık %19'u idrarda glukuronize metabolitler şeklinde atılmıştır. Kararlı durum koşullarında glukuronidlerin üriner itrahı %10'un altına düşmüştür. Feçeste bulunan ana bileşik, glukuronidlerin intestinal degradasyonundan veya M-2 metabolitinin (N-oksit) ve emilime uğramamış regorafenibin redüksiyonundan elde edilebilmiştir. Gastrointestinal kanalda mikrobiyal flora M-5 metabolitini M-4'e indirgeyerek M-4 için geri emilime (enterohepatik dolaşım) imkan verebilir. M-5 sonuç olarak feçeste M-4 üzerinden M-6 şeklinde atılmaktadır.
Doğrusallık/Doğrusal olmayan durum:
Kararlı durumda regorafenibin sistemik maruziyeti 60 mg'a kadar dozla orantılı olarak ve
60 mg'dan daha yüksek dozlarda dozla orantılı değerden daha düşük bir değerde artış göstermektedir. Kararlı durumda regorafenib birikimi plazma konsantrasyonlarında yaklaşık 2 katlık bir artışla sonuçlanır ve bu, eliminasyon yarılanma ömrü ve doz uygulama sıklığı ile tutarlıdır. Kararlı durumda, 160 mg regorafenibin oral uygulamasından sonra regorafenib yaklaşık 3,9 mg/L'lik (8,1 mikromolar) ortalama doruk plazma düzeylerine erişir ve ortalama plazma konsantrasyonlarının tepe:vadi oranı 2'den düşüktür.
Her iki metabolit de (M-2 ve M-5) doğrusal olmayan birikim göstermektedir. Tek doz regorafenibin verilmesinden sonra M-2 ve M-5'in plazma konsantrasyonları, ana bileşene oranla çok daha düşük olmakla birlikte, kararlı durumda plazma konsantrasyonları regorafenib ile benzerdir.
Hastalardaki karakteristik özellikler
Böbrek yetmezliği olan hastalar:
Mevcut klinik verilere ve fizyoloji bazlı farmakokinetik modellemeye göre, regorafenibin ve metabolitleri M-2 ve M-5'in kararlı durum maruziyeti hafif ve orta derecede böbrek yetmezliği olan hastalar ve böbrek fonksiyonları normal olan hastalarda benzerdir.
Şiddetli böbrek yetmezliği olan hastalar ile normal böbrek fonksiyonu olan hastalar karşılaştırıldığında, regorafenib maruziyeti benzerken, klinik olarak anlamlı kabul edilmeyen kararlı durum koşullarında M-2 ve M-5 maruziyeti yaklaşık %30 oranında azalmıştır.
Regorafenibin farmakokinetiği son evre böbrek yetersizliği olan hastalarda araştırılmamıştır. Bununla birlikte, fizyoloji bazlı farmakokinetik modelleme, bu hastalarda maruziyette görülebilecek herhangi bir anlamlı değişikliği öngörmemektedir.
Karaciğer yetmezliği olan hastalar:
Regorafenibin ve metabolitleri olan M-2 ve M-5'in maruziyeti, hafif derecede karaciğer yetmezliği olan (Child-Pugh A) hastalarda ve karaciğer fonksiyonları normal olan hastalarda benzerdir. 100 mg tek doz regorafenib uygulanmasından sonra orta derecede karaciğer yetmezliği (Child-Pugh B) olan hastalardaki maruziyetinin, karaciğer fonksiyonları normal olan hastalardaki farmakokinetiği ile benzer olduğuna ilişkin sınırlı veri bulunmaktadır. Child-Pugh C (şiddetli) karaciğer yetmezliği olan hastalara ilişkin bilgi bulunmamaktadır. Regorafenib başlıca karaciğer yoluyla atılmaktadır ve bu hasta popülasyonunda maruziyet artabilmektedir.
Geriyatrik hastalar:
Regorafenibin farmakokinetiği, çalışılan yaş aralığında (29 – 85 yaş), yaştan etkilenmemiştir.
Cinsiyet:
Regorafenibin farmakokinetiği cinsiyetten etkilenmemektedir.
Etnik farklılıklar:
Farklı Asya popülasyonlarındaki (Çinli, Japon, Koreli) regorafenib maruziyeti beyaz ırkta görülen ile aynı aralıktadır.
Kardiyak Elektrofizyoloji/QT uzaması:
Erkek ve kadın kanser hastalarında yapılan özel bir QT çalışmasında, kararlı durumda 160
5.3. Klinik öncesi güvenlilik verileri
Sistemik toksisite
Farelere, sıçanlara ve köpeklere tekrarlanan doz uygulanmasından sonra esas olarak böbreklerde, karaciğerde, sindirim sisteminde, kalpte, tiroid bezinde, kan ve lenf sisteminde, endokrin sistemde, üreme sisteminde ve deride olmak üzere bir dizi organda advers etkiler gözlenmiştir. Sıçanlarda 26 haftalık tekrar dozlu toksisite çalışmasında kalpte atrioventriküler damarlarda kalınlaşma insidansında hafif artış görülmüştür. Nedeni yaşa bağlı fizyolojik proseslerin hızlanması olabilir. Bu etkiler, öngörülen insan maruziyeti aralığında veya altındaki sistemik maruziyetlerde meydana gelmiştir (EAA karşılaştırmasına dayalı olarak).
Genç ve büyüme dönemindeki hayvanların ve yavru sıçanların dişlerinde ve kemiklerinde değişiklikler ve üreme sistemindeki advers etkiler daha belirgin olup, bu durum çocuklar ve ergenler için potansiyel bir riske işaret etmektedir.
Genotoksisite ve karsinojenisite
Regorafenibin karsinojenik potansiyeline ilişkin çalışma yürütülmemiştir.
Fareler üzerinde yürütülen in vitro ve in vivo standart analizlerde, regorafenibin genotoksik potansiyeline ilişkin bulgu gözlenmemiştir.
Üreme ve gelişim toksisitesi
Fertiliteye özgü çalışmalar yürütülmemiştir. Bununla birlikte, sıçanlarda ve köpeklerde öngörülen insan maruziyeti altındaki maruziyetlerde (EAA karşılaştırmasına dayalı olarak) tekrarlanan doz uygulamasını takiben testislerde, overlerde ve uterusta gözlenen morfolojik değişikliklere dayalı olarak regorafenibin erkek ve dişi üreme sistemini advers olarak etkileme potansiyelinin olduğu dikkate alınmalıdır. Gözlenen değişiklikler sadece kısmen geri dönüşümlüdür.
Tavşanlarda, öngörülen insan maruziyetinin altındaki maruziyetlerde (EAA karşılaştırmasına dayalı olarak) regorafenibin intrauterin gelişim üzerindeki etkisi gösterilmiştir. Başlıca bulgular üriner sistem, kalp ve majör damarlar ve iskelette malformasyonları içermiştir.
Çevresel Risk Değerlendirme (ÇRD)
Çevresel risk değerlendirme çalışmaları, regorafenibin çevreye karşı kalıcı, biyoakümülatif ve toksik olma potansiyeline sahip olduğunu; yerüstü suyu ve çökelti kısımlarına risk oluşturabildiğini göstermiştir.
 Sırt Ağrısı
Sırt ağrısı birden bire ortaya
çıkıp şiddetli (akut) olabilir veya zamanla gelişip daha uzun
süreli sorunlara (kronik) neden olabilir.
Sırt Ağrısı
Sırt ağrısı birden bire ortaya
çıkıp şiddetli (akut) olabilir veya zamanla gelişip daha uzun
süreli sorunlara (kronik) neden olabilir. |
 İnme
İnme, beynin hasar görmesinin sonucudur. Bu hasar, beynin bir kısmındaki ya bir kanama
ya da akut kan eksikliği nedeniyle o kısmın geçici ya da kalıcı olarak işlevini yapamamasına
yol açar.
İnme
İnme, beynin hasar görmesinin sonucudur. Bu hasar, beynin bir kısmındaki ya bir kanama
ya da akut kan eksikliği nedeniyle o kısmın geçici ya da kalıcı olarak işlevini yapamamasına
yol açar. |
İLAÇ GENEL BİLGİLERİ
Bayer Türk Kimya San. Tic. Ltd. Şti.
| Geri Ödeme Kodu | A15618 |
| Satış Fiyatı | 73173.5 TL [ 9 Jun 2026 ] |
| Önceki Satış Fiyatı | 73173.5 TL [ 22 May 2026 ] |
| Original / Jenerik | Original İlaç |
| Reçete Durumu | Normal Reçeteli bir ilaçdır. |
| Barkodu | 8699546090303 |
| Etkin Madde | Regorafenib |
| ATC Kodu | L01EX05 |
| Birim Miktar | 40 |
| Birim Cinsi | MG |
| Ambalaj Miktarı | 84 |
| Antineoplastik ve İmmünomodülatör Ajanlar |
| İthal ( ref. ülke : Fransa ) ve Beşeri bir ilaçdır. |
İLAÇ EŞDEĞERLERİ
| Eşdeğer İlaç Adı | Barkodu | İlaç Fiyatı |
|---|---|---|
| Eşdeğer bir ilaç bulunamadı |
 Kanseri") |
Rahim Boyu ( Serviks ) Kanseri Rahim boynu (serviks) kanseri 35 yaş altı kadınlarda görülen vakalarda meme kanserinden sonra ikinci sırayı alır.Serviks kanserinin gelişmesi yıllarca sürebilir. |
 |
Doğum Sonrası Depresyonu Doğum sonrası depresyonu, doğumdan sonra her on kadından biri tarafından tecrübe edilen stresli bir durumdur. |
 |
Ağız Kanseri Ağız kanserinin en yaygın türleri, dudak, dil, dişetidir. Nadiren yanak içi veya damak bölgelerini de içine alır. |