STIVARGA 40 mg 84 film kapl� tablet K�sa �r�n Bilgisi
{ Regorafenib }
1. BE�ER� TIBB� �R�N�N ADI
STIVARGA® 40 mg film kapl� tablet Sitotoksik
2. KAL�TAT�F VE KANT�TAT�F B�LE��M
Etkin madde
Regorafenib 40 mg (41,49 mg regorafenib monohidrat olarak)
Yard�mc� maddeler
STIVARGA'n�n g�nl�k dozunda (4 tablet; 160 mg);
Sodyum 2,438 mmol (56,06 mg'a e�de�er)
Lesitin (soyadan elde edilir) 1,68 mg Yard�mc� maddeler i�in 6.1'e bak�n�z.
3. FARMAS�T�K FORMU
Film kapl� tablet.
Bir y�z�nde “BAYER” logosu, di�er y�z�nde “40” i�areti bulunan, 16 mm uzunlu�unda ve 7 mm kal�nl���nda, a��k pembe renkte oval film kapl� tabletler.
4. KL�N�K �ZELL�KLER
4.1. Terap�tik endikasyonlar
STIVARGA, �ncesinde floropirimidin-, oksaliplatin- ve irinotekan bazl� kemoterapi, anti- VEGF (anti vask�ler endotelyal b�y�me fakt�r�) tedavisi ve RAS-do�al tip ise anti-EGFR (anti epidermal b�y�me fakt�r� resept�r�) ile tedavi g�rm�� ve progresyon g�stermi�, ECOG performans skoru: 0-1 olan, yeterli organ fonksiyonu bulunan ve ya�am beklentisi �� aydan fazla olan, rezeke edilemeyen metastatik kolorektal kanserli (mKRK) hastalarda endikedir.
4.2. Pozoloji ve uygulama �ekli
STIVARGA antikanser tedavisinin uygulanmas�nda deneyimli hekimler taraf�ndan re�ete
edilmelidir.
STIVARGA ile 4 haftal�k bir tedavi k�r� ila� al�nan 3 tedavi haftas� ve ard�ndan tedavisiz 1 haftadan olu�ur. STIVARGA'n�n �nerilen dozu 3 tedavi haftas� s�resince g�nde bir kez oral yolla al�nan 160 mg regorafenibdir (40 mg regorafenib i�eren 4 adet STIVARGA tablet).
Tedaviye yarar sa�land��� s�rece veya tedavi s�ras�nda kabul edilemeyen toksisite ortaya
��kana kadar devam edilmelidir (bkz. B�l�m 4.4).
Performans skoru (PS) 2 veya daha y�ksek olan hastalar klinik �al��maya dahil edilmemi�tir. PS≥2'den b�y�k olan hastalar i�in k�s�tl� veri bulunmaktad�r.
Doz modifikasyonlar�
Bireysel g�venlili�e ve tolerabiliteye ba�l� olarak ilac�n kullan�m�na ara verilmesi ve/veya dozun azalt�lmas� gerekebilir. Doz modifikasyonlar� 40 mg'l�k (bir tablet) doz ad�mlar� �eklinde uygulan�r. En d���k �nerilen g�nl�k doz 80 mg'd�r. Maksimum g�nl�k doz 160 mg'd�r.
El-ayak deri reaksiyonlar� (EADR)/ palmar-plantar eritrodizestezi sendromu) durumunda
�nerilen doz modifikasyonlar� ve �nlemler Tablo 1'de �zetlenmi�tir.
Tablo 1: EADR durumunda �nerilen doz modifikasyonlar� ve �nlemler
Deri toksisitesi derecesi | Ortaya ��k�� | �nerilen doz modifikasyonlar� ve �nlemler |
Derece 1 | Herhangi bir zaman | Mevcut doz d�zeyi korunur ve semptomlar�n giderilmesi i�in derhal destekleyici �nlemler al�n�r. |
Derece 2 | �lk olay | Doz 40 mg (bir tablet) azalt�l�r ve derhal destekleyici �nlemler al�n�r.
Dozun azalt�lmas�na kar��n iyile�me g�r�lmezse, en az 7 g�n s�reyle, toksisite Derece 0-1'e gerileyene kadar tedaviye ara verilir.
Tedaviden sorumlu hekimin karar� do�rultusunda dozun yeniden art�r�lmas�na izin verilir. |
7 g�n i�inde iyile�me olmamas� veya ikinci kez ortaya ��k�� | Toksisite Derece 0-1'e gerileyene kadar tedaviye ara verilir.
Aradan sonra tedaviye yeniden ba�larken, doz 40 mg (bir tablet) azalt�l�r.
Tedaviden sorumlu hekimin karar� do�rultusunda dozun yeniden art�r�lmas�na izin verilir. | |
���nc� kez ortaya ��k�� | Toksisite Derece 0-1'e gerileyene kadar tedaviye ara verilir.
Aradan sonra tedaviye yeniden ba�larken, doz 40 mg (bir tablet) azalt�l�r.
Tedaviden sorumlu hekimin karar� do�rultusunda dozun yeniden art�r�lmas�na izin verilir. | |
D�rd�nc� kez ortaya ��k�� | STIVARGA ile tedavi tamamen sonland�r�l�r. | |
Derece 3
| �lk olay
| Derhal destekleyici �nlemler al�n�r. En az 7 g�n s�reyle, toksisite Derece 0-1'e gerileyene kadar tedaviye ara verilir. |
|
| (bir tablet) azalt�l�r.
Tedaviden sorumlu hekimin karar� do�rultusunda dozun yeniden art�r�lmas�na izin verilir. |
�kinci kez ortaya ��k�� | Derhal destekleyici �nlemler al�n�r. En az 7 g�n s�reyle, toksisite Derece 0-1'e gerileyene kadar tedaviye ara verilir.
Aradan sonra tedaviye yeniden ba�larken, doz 40 mg (bir tablet) azalt�l�r. | |
���nc� kez ortaya ��k�� | STIVARGA ile tedavi tamamen sonland�r�l�r. |
Karaci�er fonksiyon testlerinde, STIVARGA tedavisi ile ili�kili oldu�u d���n�len k�t�le�menin g�zlendi�i durumlarda �nerilen �nlemler ve doz modifikasyonlar� Tablo 2'de �zetlenmi�tir (ayr�ca bkz. B�l�m 4.4).
Tablo 2: �la�la ili�kili karaci�er fonksiyon testi anormallikleri durumunda �nerilen �nlemler ve doz modifikasyonlar�
ALT ve/veya AST de�erlerinde g�zlenen art��lar | Ortaya ��k�� | �nerilen �nlemler ve doz modifikasyonlar� |
≤5 x Normalin �st s�n�r� (N�S) (en fazla Derece 2) | Herhangi bir zamanda ortaya ��k�� | STIVARGA tedavisine devam edilir. Transaminazlar <3 x N�S (Derece 1) veya ba�lang�� de�erine d�nene kadar haftada bir kez karaci�er fonksiyonlar� takip edilir. |
>5 x N�S ila ≤20 x N�S (Derece 3) | �lk olay | STIVARGA tedavisine ara verilir. Transaminazlar <3 x N�S veya ba�lang�� de�erine d�nene kadar haftada bir kez takip edilir.
Yeniden ba�lama: Potansiyel yarar hepatotoksisite riskine a��r bas�yorsa, STIVARGA tedavisi yeniden ba�lat�l�r. Aradan sonra tedaviye yeniden ba�larken, doz 40 mg (bir tablet) azalt�l�r ve en az 4 hafta boyunca haftada bir kez karaci�er fonksiyonlar� takip edilir. |
N�ks | STIVARGA ile tedavi tamamen sonland�r�l�r. | |
>20 x N�S (Derece 4) | Herhangi bir zamanda ortaya ��k�� | STIVARGA ile tedavi tamamen sonland�r�l�r. |
>2 x N�S bilirubin ile | Herhangi bir | STIVARGA ile tedavi tamamen sonland�r�l�r. |
e� zamanl� olarak | zamanda ortaya |
|
>3 x N�S | ��k�� | Karaci�er fonksiyonlar� d�zelene veya |
(Derece 2 veya daha |
| ba�lang��taki duruma d�nene kadar haftada |
y�ksek) |
| bir kez takip edilir. |
|
| �stisna: Gilbert sendromu olan ve transaminaz |
| d�zeyleriy�kselen hastalar, ALT ve/veya ASTde�erlerindeart��larg�zlendi�inde |
|
| yukar�da �zetlenen �nerilere g�re tedavi edilmelidir. |
ALT: Alanin aminotransferaz AST: Aspartat aminotransferaz
Uygulama �ekli:
STIVARGA, oral kullan�m i�indir.
STIVARGA her g�n ayn� saatte al�nmal�d�r. Tabletler %30'dan d���k oranda ya� i�eren hafif bir yeme�in ard�ndan bir b�t�n halinde su ile yutulmal�d�r (bkz. B�l�m 5.2). Hafif (d���k ya� i�eren) bir ���ne �rnek olarak; bir fincan tah�l (yakla��k 30 g), bir bardak ya�s�z s�t, bir dilim re�elli k�zarm�� ekmek, 1 bardak elma suyu ve bir fincan kahve veya �ay verilebilir (520 kalori, 2 g ya�).
Hasta STIVARGA'n�n bir dozunu almay� unutursa, unuttu�u dozu ayn� g�n i�inde hat�rlar hat�rlamaz almal�d�r. Hasta unuttu�u dozu dengelemek i�in ayn� g�n �ift doz almamal�d�r.
Regorafenib uygulanmas�ndan sonra hastada kusma g�zlenirse, hasta ilave tablet
almamal�d�r.
�zel pop�lasyonlara ili�kin ek bilgiler:
B�brek yetmezli�i:
Mevcut klinik veriler hafif, orta veya �iddetli b�brek yetmezli�i bulunan hastalarda regorafenib ve metabolitleri M-2 ile M-5 i�in maruziyetin b�brek fonksiyonu normal olan hastalardaki maruziyete benzer oldu�unu g�stermektedir. Hafif, orta veya �iddetli b�brek yetmezli�i olan hastalarda doz ayarlamas� gerekli de�ildir (Bkz. B�l�m 5.2 Farmakodinamik �zellikler).
Karaci�er yetmezli�i:
Regorafenib ba�l�ca karaci�er yoluyla at�lmaktad�r.
Klinik �al��malarda, hafif karaci�er yetmezli�i (Child-Pugh A) olan hastalar ve karaci�er fonksiyonlar� normal olan hastalar aras�nda maruziyet, g�venlilik veya etkililik a��s�ndan anlaml� farkl�l�klar g�zlenmemi�tir. Hafif karaci�er yetmezli�i olan hastalar i�in doz ayarlamas� gerekli de�ildir. Orta derecede (Child-Pugh B) karaci�er yetmezli�i olan hastalarda sadece k�s�tl� veri bulundu�undan, doz �nerisi sa�lanamamaktad�r. Bu hastalarda genel g�venlili�in yak�ndan takip edilmesi �nerilmektedir (ayr�ca bkz. B�l�m 4.4 ve B�l�m 5.2).
STIVARGA �iddetli karaci�er yetmezli�i (Child-Pugh C) olan hasta pop�lasyonunda ara�t�r�lmad���ndan, STIVARGA'n�n bu hastalarda kullan�lmas� �nerilmemektedir.
Pediyatrik pop�lasyon:
STIVARGA'n�n metastatik kolorektal kanser endikasyonunda pediyatrik pop�lasyonda
kullan�m� bulunmamaktad�r.
STIVARGA'n�n gastrointestinal stromal t�m�r (G�ST) endikasyonunda �ocuklarda ve 18 ya��n alt�ndaki ergenlerde g�venlili�i ve etkilili�i belirlenmemi�tir. Veri bulunmamaktad�r.
STIVARGA'n�n hepatosel�ler karsinom endikasyonunda pediyatrik pop�lasyonda kullan�m�
bulunmamaktad�r.
Geriyatrik pop�lasyon:
Klinik �al��malarda, ya�l�lar (65 ya� ve �zeri) ve daha gen� hastalar aras�nda maruziyet, g�venlilik veya etkililik a��s�ndan anlaml� farkl�l�klar g�zlenmemi�tir. Ya�l� hastalarda doz ayarlamas� gerekli de�ildir (ayr�cabkz.B�l�m5.2).
Cinsiyet:
Klinik �al��malarda, erkek ve kad�n hastalar aras�nda maruziyet, g�venlilik veya etkililik a��s�ndan anlaml� farkl�l�klar g�zlenmemi�tir. Cinsiyete g�re doz ayarlamas� gerekli de�ildir (ayr�ca bkz. B�l�m 5.2).
Etnik farkl�l�klar:
Klinik �al��malarda, farkl� etnik gruplara ait hastalar aras�nda maruziyet, g�venlilik veya etkililik a��s�ndan anlaml� farkl�l�klar g�zlenmemi�tir. Etnik k�kene g�re doz ayarlamas� gerekli de�ildir (ayr�ca bkz. B�l�m 5.2). El ayak deri reaksiyonu (EADR), karaci�er fonksiyon testlerinde ciddi anormallikler ve karaci�er fonksiyon bozuklu�u insidans�n�n STIVARGA tedavisi alan Asyal� (�zellikle Japon) hastalarda beyaz �rka k�yasla daha y�ksek oldu�u g�zlenmi�tir. Klinik �al��malarda STIVARGA tedavisi alan Asyal� hastalar�n primer olarak Do�u Asya k�kenli oldu�u kaydedilmi�tir.
4.3. Kontrendikasyonlar
STIVARGA, etkin madde ya da yard�mc� maddelerin herhangi birine (bkz.B�l�m 6.1) kar�� a��r� duyarl�l��� olan ki�ilerde kontrendikedir.
E�er f�st�k ya da soya alerjisi varsa STIVARGA kullan�lmamal�d�r.
4.4. �zel kullan�m uyar�lar� ve �nlemleri
Anevrizmalar ve arter diseksiyonlar�
VEGF yolak inhibit�rlerinin, hipertansiyonu olan veya olmayan hastalarda kullan�lmas�, anevrizmalar ve/veya arter diseksiyonlar� olu�umunu kolayla�t�rabilir. STIVARGA'ya ba�lamadan �nce hipertansiyon veya anevrizma �yk�s� gibi risk fakt�rleri olan hastalarda bu risk dikkatle de�erlendirilmelidir.
Karaci�er �zerine etkiler:
STIVARGA ile tedavi edilen hastalarda s�kl�kla karaci�er fonksiyon testlerinde (alanin amino transferaz [ALT], aspartat aminotransferaz [AST] ve bilirubin) anormallikler g�zlenmi�tir. K���k bir hasta grubunda �iddetli karaci�er fonksiyon testi anormallikleri (Derece 3 ila 4) ve klinik seyir g�steren karaci�er fonksiyon bozuklu�u (�l�mc�l sonu�lar dahil) bildirilmi�tir (bkz. B�l�m 4.8). Klinik �al��malarda karaci�er fonksiyon testlerinde ciddi anormallik ve karaci�er fonksiyon bozuklu�u insidans�n�n STIVARGA tedavisi alan Asyal� (�zellikle Japon) hastalarda beyaz �rka k�yasla daha y�ksek oldu�u g�zlenmi�tir (bkz. B�l�m 4.2).
STIVARGA ile tedaviye ba�lamadan �nce karaci�er fonksiyon testlerinin (ALT, AST ve bilirubin) yapt�r�lmas� ve bu de�erlerin tedavinin ilk 2 ay� boyunca yak�ndan takip edilmesi (en az iki haftada bir) �nerilmektedir. Bu d�nemin ard�ndan ise, periyodik takibe en az ayda bir ve klinik olarak gerekli oldu�unda devam edilmelidir.
Regorafenib bir �ridin difosfat glukuronozil transferaz (UGT) 1A1 inhibit�r�d�r (bkz. B�l�m 4.5). Gilbert sendromu olan hastalarda hafif, indirekt (konjuge olmayan) hiperbilirubinemi g�r�lebilir.
Karaci�er fonksiyon testlerinde, STIVARGA tedavisi ile ili�kili oldu�u d���n�len k�t�le�menin g�zlendi�i hastalar i�in (post-hepatik kolestaz veya hastal�k progresyonu gibi alternatif bir nedenin olmad��� durumlarda) Tablo 2'de verilen doz modifikasyonlar� ve takip �nerileri izlenmelidir (bkz. B�l�m 4.2).
Regorafenib, ba�l�ca karaci�eryoluylaelimineedilmektedir. Hafif veya orta derecede
�nerilmektedir (bkz. B�l�m 4.2 B�l�m 5.2). STIVARGA, �iddetli karaci�er yetmezli
(Child-Pugh C) olan hasta pop�lasyonunda ara�t�r�lmad���ndan ve bu hastalarda maruziyet daha y�ksek olabilece�inden STIVARGA'n�n bu hastalarda kullan�lmas� �nerilmemektedir.
Enfeksiyonlar:
STIVARGA enfeksiyon olaylar�n�n insidans�nda art�� ile ili�kilendirilmi�tir ve bu olaylar�n bir k�sm� �l�mc�l olmu�tur (bkz. B�l�m 4.8).
Enfeksiyon olaylar�n�n k�t�le�mesi halinde, STIVARGA tedavisine ara verilmesi
d���n�lmelidir.
Hemoraji:
STIVARGA, baz�lar� �l�mc�l olan hemorajik olaylar�n insidans�nda art�� ile ili�kilendirilmi�tir (bkz. B�l�m 4.8). Kanamaya yatk�nl�k yaratan durumlar�n g�r�ld���, antikoag�lanlar (�rn. varfarin ve fenprokumon) veya kanama riskini art�ran di�er t�bbi �r�nlerle e�zamanl� tedavi edilen hastalar�n kan say�mlar� ve koag�lasyon parametreleri takip edilmelidir. Karaci�er sirozu bulunan hastalarda, STIVARGA tedavisine ba�lanmadan �nce standart olarak �zofageal varisler i�in tarama ve ard�ndan tedavi uygulanmal�d�r. Acil t�bbi giri�im gerektiren �iddetli kanama durumunda, STIVARGA'n�n tamamen kesilmesi d���n�lmelidir.
Gastrointestinal perforasyon ve fist�l:
STIVARGA ile tedavi edilen hastalarda gastrointestinal perforasyon (fatal sonu�lar� olanlar dahil) ve fist�l olu�umu bildirilmi�tir (bkz. B�l�m 4.8). Bu olaylar�n ayn� zamanda intra- abdominal maligniteleri olan hastalarda hastal�kla ili�kili yayg�n komplikasyonlar olduklar� bilinmektedir. Gastrointestinal perforasyon veya fist�l olu�an hastalarda STIVARGA'n�n kesilmesi �nerilmektedir.
Kardiyak iskemi ve infarkt�s:
STIVARGA, miyokard iskemisi ve infarkt�s� insidans�nda art��la ili�kilendirilmi�tir (bkz. B�l�m 4.8). Stabil olmayan anjina veya yeni ba�lang��l� anjina (STIVARGA tedavisine ba�lamadan �nceki 3 ay i�inde), yak�n zamanda miyokard enfarkt�s� ge�irmi� (STIVARGA tedavisine ba�lamadan �nceki 6 ay i�inde) ve New York Kalp Derne�i (NYHA) s�n�flamas�na g�re kalp yetmezli�i 2 veya daha y�ksek olan hastalar klinik �al��malara dahil edilmemi�tir.
�skemik kalp hastal��� �yk�s� olan hastalar miyokard iskemisinin klinik bulgu ve semptomlar� a��s�ndan takip edilmelidir. Kardiyak iskemi ve/veya infarkt�s� geli�en hastalarda iyile�me g�r�lene kadar STIVARGA tedavisine ara verilmelidir.
STIVARGA ile tedaviye yeniden ba�lama karar� al�n�rken, her bir hasta i�in potansiyel yarar ve risklere ili�kin dikkatli bir inceleme yap�lmal�d�r. Hastada d�zelme g�r�lmezse STIVARGA tamamen kesilmelidir.
Geri D�n���ml� Posterior L�koensefalopati Sendromu (PRES):
STIVARGA tedavisi ile ili�kili olarak Geri d�n���ml� posterior l�koensefalopati sendromu
(GPLS) bildirilmi�tir (bkz. B�l�m 4.8).
GPLS'nin bulgu ve semptomlar� aras�nda n�bet, ba� a�r�s�, mental durumda de�i�iklikler, konf�zyon, hipertansiyon ile ili�kili olan ya da olmayan g�rme bozukluklar� veya kortikal k�rl�k yer almaktad�r. GPLS tan�s�n�n beyin g�r�nt�lemesi ile do�rulanmas� gerekmektedir. GPLS geli�en hastalarda, hipertansiyon kontrol� ve di�er semptomlar i�in destekleyici t�bbi kontrol ile birlikte STIVARGA'n�n kesilmesi �nerilmektedir.
Arteriyel hipertansiyon:
STIVARGA arteriyel hipertansiyon insidans�nda art��la ili�kilendirilmi�tir (bkz. B�l�m 4.8). Hipertansiyon geli�en �o�u hastada, hipertansiyon ba�lang�c� STIVARGA tedavisinin ilk siklusunda ortaya ��km��t�r. Kan bas�nc� STIVARGA ile tedaviye ba�lamadan �nce kontrol edilmelidir. Kan bas�nc� izlenmeli ve standart t�bbi uygulamalar do�rultusunda hipertansiyon tedavi edilmelidir. Yeterli t�bbi tedaviye kar��n �iddetli veya diren�li hipertansiyon vakalar�nda, tedaviden sorumlu hekimin karar�na ba�l� olarak STIVARGA tedavisine ge�ici olarak ara verilmeli ve/veya doz azalt�lmal�d�r (bkz. B�l�m 4.2). Hipertansif kriz durumunda STIVARGA kullan�m� sonland�r�lmal�d�r.
Yara iyile�me komplikasyonlar� :
Anti-anjiyojenik �zellikleri bulunan t�bbi �r�nler yara iyile�mesini geciktirebilece�i veya engelleyebilece�i i�in, b�y�k bir ameliyat ge�irecek olan hastalarda STIVARGA kullan�m�n�n �nlem olarak ge�ici �ekilde kesilmesi �nerilir. B�y�k bir ameliyattan sonra STIVARGA ile tedaviye devam etme karar�, yeterli yara iyile�mesine dair klinik de�erlendirmelere dayanmal�d�r.
Dermatolojik toksisite:
El-ayak deri reaksiyonu (EADR /palmar-plantar eritrodizestezi sendromu) ve d�k�nt� STIVARGA ile g�zlenen en yayg�n dermatolojik advers ila� reaksiyonlar�n� temsil etmektedir (bkz. B�l�m 4.8). Klinik �al��malarda STIVARGA ile tedavi edilen Beyaz hastalara k�yasla Asyal� hastalarda (�zellikle Japon) EADR insidans�n�n daha y�ksek oldu�u g�zlenmi�tir (bkz. B�l�m 4.2). EADR'yi �nlemek i�in al�nacak �nlemler aras�nda nas�r kontrol� ve ayak tabanlar�ndaki ve avu� i�lerindeki bas�nc� azaltmak �zere ayakkab� yast�klar�n�n ve eldivenlerin kullan�lmas� yer almaktad�r.
EADR tedavisi, semptomlar�n giderilmesi i�in keratolitik kremlerin (�rn., sadece etkilenmi� alanlara az miktarda uygulanan �re-, salisilik asit- veya alfa hidroksil asit bazl� kremler) ve nemlendirici kremlerin (istenildi�i kadar uygulanan) kullan�lmas�n� i�erebilir. STIVARGA dozunun azalt�lmas� ve/veya tedaviye ge�ici olarak ara verilmesi ya da �iddetli veya diren�li durumlarda STIVARGA'n�n tamamen kesilmesi d���n�lmelidir (bkz. B�l�m 4.2).
Biyokimyasal ve metabolik laboratuvar testi anormallikleri:
STIVARGA, elektrolit anormalliklerinin (hipofosfatemi, hipokalsemi, hiponatremi ve hipokalemi) ve metabolik anormalliklerinin (tiroid stim�lan hormon, lipaz ve amilazda art��lar dahil) insidans�nda art��la ili�kilendirilmi�tir. Anormallikler genellikle hafif ila orta �iddette olup, klinik seyir g�stermez ve genellikle dozun kesilmesini veya azalt�lmas�n� gerektirmez. STIVARGA tedavisi s�ras�nda biyokimyasal ve metabolik parametrelerin takip edilmesi ve gerekirse standart klinik uygulamaya g�re uygun replasman tedavisinin ba�lat�lmas� �nerilmektedir. Diren�li veya tekrar eden anlaml� anormalliklerin g�r�lmesi durumunda dozun kesilmesi veya azalt�lmas� ya da STIVARGA'n�n tamamen b�rak�lmas� d���n�lmelidir (bkz. B�l�m 4.2).
Trombotik mikroanjiyopati (TMA):
Trombotik trombositopenik purpura (TTP) dahil olmak �zere trombotik mikroanjiyopati (TMA), regorafenib kullan�m�yla ili�kilendirilmi�tir (bkz. b�l�m 4.8). Hemolitik anemi, trombositopeni, yorgunluk, de�i�ken n�rolojik belirtiler, b�brek yetmezli�i ve ate� ile ba�vuran hastalarda TMA tan�s� d���n�lmelidir. TMA geli�en hastalarda regorafenib tedavisi kesilmelidir ve acil tedavi gereklidir. Tedavinin kesilmesinden sonra TMA'n�n etkilerinin tersine d�nd��� g�zlemlenmi�tir.
Hastal��a �zg� �nlemler - Hepatosel�ler karsinom (HSK):
Plasebo kontroll� pivot faz III �al��mada, hastalar �nceden sorafenib tedavisi alm��t�r.
Sodyum:
STIVARGA'n�n �nerilen g�nl�k dozu (4 tablet; 160 mg) 2,438 mmol (56,06 mg'a e�de�er) sodyum ihtiva eder. Bu durum, kontroll� sodyum diyetinde olan hastalar i�in g�z �n�nde bulundurulmal�d�r.
Lesitin:
STIVARGA'n�n �nerilen g�nl�k dozu (4 tablet; 160 mg), 1,68 mg lesitin (soyadan elde edilir) ihtiva eder. E�er f�st�k ya da soya alerjisi varsa STIVARGA kullan�lmamal�d�r.
4.5. Di�er t�bbi �r�nler ile etkile�imler ve di�er etkile�im �ekilleri
CYP3A4 ve UGT1A9 inhibit�rleri/CYP3A4 ind�kleyicileri:
�n vitro veriler regorafenibin, sitokrom CYP3A4 ve �ridin difosfat glukuronozil transferaz
UGT1A9 ile metabolize oldu�unu g�stermektedir.
G��l� bir CYP3A4 inhibit�r� olan ketokonazol�n (18 g�n boyunca 400 mg) tek doz regorafenib (5. g�nde 160 mg) ile birlikte uygulanmas�, regorafenibe ortalama maruziyette (EAA) yakla��k %33'l�k bir art�� ve aktif metabolitler, M-2 (N-oksit) ve M-5'e (N-oksit ve N-destemil) ortalama maruziyette yakla��k %90 azalma ile sonu�lanm��t�r. Regorafenibin ve metabolitlerinin (M-2 ve M-5) kararl� durum maruziyetleri �zerine etkileri ara�t�r�lmam�� oldu�undan, CYP3A4 aktivitesinin g��l� inhibit�rlerinin (�rn., klaritromisin, greyfurt suyu, itrakonazol, ketokonazol, posakonazol, nefazodon, telitromisin ve vorikonazol) STIVARGA ile birlikte kullan�lmas�ndan ka��n�lmas� �nerilmektedir.
Regorafenib ve onun metabolitlerinin kararl� durum maruziyetine etkileri �al���lmad���ndan; regorafenib tedavisi s�resince, g��l� UGT1A9 inhibit�rlerinin (�rn. mefenamik asit, diflunisal ve niflumik asit) birlikte kullan�m�ndan ka��n�lmal�d�r.
G��l� bir CYP3A4 ind�kleyicisi olan rifampisinin (9 g�n boyunca 600 mg) tek doz regorafenib (7. g�nde 160 mg) ile birlikte uygulanmas�, regorafenibe ortalama maruziyette (EAA) yakla��k %50 azalma ve aktif metabolit M-5'e ortalama maruziyette 3- ila 4- kat art�� ile sonu�lan�rken, aktif metabolit M-2'ye maruziyette herhangi bir de�i�iklik g�zlenmemi�tir. Di�er g��l� CYP3A4 ind�kleyicileri de (�rn., fenitoin, karbamazepin, fenobarbital, St.John's Wort) regorafenibin metabolizmas�n� art�rabilirler. Regorafenibin plazma konsantrasyonundaki d���� etkilili�in azalmas�na neden olabilece�inden, g��l� CYP3A4 ind�kleyicilerinin kullan�m�ndan ka��n�lmal� veya CYP3A4'� ind�kleme potansiyeli olmayan ya da �ok d���k bir potansiyele sahip e�zamanl� kullan�lacak alternatif bir t�bbi �r�n�n se�ilmesi d���n�lmelidir.
Meme Kanseri Diren� Proteini (BCRP) ve P-glikoprotein substratlar�:
BCRP substrat� olan rosuvastatin tek doz (5 mg) uygulamas�ndan �nce regorafenib (14 g�n 160 mg) uygulamas�, ortalama rosuvastatin maruziyetinde (EAA) 3,8 kat art�� ile Cde�erinde 4,6 kat art��a sebep olmu�tur.
Klinik veriler, regorafenibin digoksinin farmakokinetik �zellikleri �zerinde etkisi olmad���n� bu sebeple klinik olarak anlaml� ila� etkile�imleri olmadan digoksin gibi p-glikoprotein substratlar� ile e�zamanl� uygulanabilece�ini g�stermi�tir.
UGT1A1 ve UGT1A9 substratlar�:
�n vitro veriler, gerek regorafenibin gerekse aktif metaboliti olan M-2'nin, in vivo kararl� durumda elde edilen konsantrasyonlarda, �ridin difosfat glukuronil transferazlar UGT1A1 ve UGT1A9'un arac�l�k etti�i glukuronidasyonu inhibe etti�ini g�stermektedir. Di�er yandan M- 5 sadece UGT1A1'i inhibe etmektedir.
�rinotekan uygulanmas�ndan �nce 5 g�nl�k bir ara ile regorafenib uygulanmas�, UGT1A1
substrat� ve irinotekan aktif metaboliti olan SN-38'e ortalama maruziyette (EAA) yakla��k
%44'l�k art��la sonu�lanm��t�r. Ayn� zamanda irinotekan�n EAA'da yakla��k %28'lik art��� g�zlenmi�tir. Bu durum, regorafenibin UGT1A1 ve UGT1A9 substratlar� ile birlikte uygulanmas�n�n sistemik maruziyeti art�rabilece�ini g�stermektedir. Bu bulgular�n klinik anlam� bilinmemektedir.
Meme Kanseri Diren� Proteini (BCRP) ve P-glikoprotein substratlar� inhibit�r/ind�kleyicileri:
�n vitro veriler M-2 ve M-5 aktif metabolitlerinin BCRP ve P-glikoproteinin substratlar� oldu�unu g�stermektedir. BCRP ve P-glikoproteinin inhibit�rleri ve ind�kleyicilerinin M-2 ve M-5 maruziyetini etkileyebilir. Bu bulgular�n klinik �nemi bilinmemektedir (bkz. B�l�m 5.2)
CYP izoform-selektif substratlar:
�n vitro veriler regorafenibin, in vivo kararl� durumda elde edilen konsantrasyonlarda (8,1 mikromolar doruk plazma konsantrasyonu) sitokrom CYP2C8 (Kde�eri 0,6 mikromolar), CYP2C9 (Kde�eri 4,7 mikromolar), CYP2B6 (Kde�eri 5,2 mikromolar)'n�n yar��mal� inhibit�r� oldu�unu g�stermektedir. CYP3A4 (Kde�eri 11,1 mikromolar) ve CYP2C19 (16,4 mikromolar Kde�eri)'a kar�� in vitro inhibe edici potens daha az belirgindir.
160 mg regorafenibin 14 g�n s�reyle uygulanmas�n�n CYP2C8 (rosiglitazon), CYP2C9 (S- varfarin), CYP2C19 (omeprazol) ve CYP3A4 (midazolam) prob substratlar�n�n farmakokineti�i �zerindeki etkisini de�erlendirmek �zere klinik prob substrat �al��mas� yap�lm��t�r.
Farmakokinetik veriler, regorafenibin, klinik olarak anlaml� bir ila� etkile�imi olmaks�z�n CYP2C8, CYP2C9, CYP3A4 ve CYP2C19 substratlar� ile e�zamanl� verilebilece�ini g�stermektedir (ayr�ca bkz. B�l�m 4.4).
Antibiyotikler:
Konsantrasyon-zaman profili, regorafenib ve metabolitlerinin enterohepatik dola��ma girebildi�ini g�stermektedir (bkz. B�l�m 5.2). Gastrointestinal mikroflora eradikasyonu i�in kullan�lan ve zay�f emilime u�rayan antimikrobiyal bir ila� olan neomisin (regorafenibin enterohepatik dola��m�yla etkile�ebilir) ile birlikte uygulama regorafenib maruziyetini etkilememi� ancak in vitro ve in vivo ko�ullarda regorafenibin farmakolojik aktivitesine benzer aktivite g�steren M-2 ve M-5 metabolitlerinin maruziyetinde yakla��k %80 azalmaya neden olmu�tur. Neomisin etkile�iminin klinik a��dan anlaml�l��� bilinmemekle birlikte,
regorafenibin etkilili�ini azalmaola s�l���vard�r.Di�erantibiyotiklerle farmakokinetik
Safra asidi ba�lay�c� ajanlar
Regorafenib, M-2 ve M-5 enterohepatik dola��m�na ge�mesi muhtemeldir (bkz. B�l�m 5.2). Kolestiramin ve kolosevelam gibi safra tuzu ba�lay�c� ajanlar, absorpsiyonu (veya reabsorpsiyonu) etkileyebilen ve bu nedenle potansiyel azalm�� maruziyet ile sonu�lanan ��z�nmeyen kompleksler olu�turarak, regorafenib ile etkile�ime girebilirler. Bu potansiyel etkile�imlerin klinik �nemi bilinmemektedir, ancak regorafenibin etkilili�inde azalmaya neden olabilir.
4.6. Gebelik ve laktasyon
Gebelik kategorisi D'dir.
�ocuk do�urma potansiyeli bulunan kad�nlar/Do�um kontrol� (Kontrasepsiyon)
�ocuk do�urma potansiyeli bulunan kad�nlar regorafenibin, fet�s �zerinde zararl� etkilere neden olabilece�i konusunda bilgilendirilmelidir.
�ocuk do�urma potansiyeli olan kad�nlar ve ayr�ca erkekler tedavi s�resince ve tedavinin tamamlanmas�n� takiben 8 haftaya kadar etkili do�um kontrol y�ntemi kullanmal�d�rlar.
Gebelik d�nemi
Regorafenibin gebe kad�nlarda kullan�m�na ili�kin bilgi mevcut de�ildir. Regorafenibin etki mekanizmas�na dayal� olarak, gebelik d�neminde uyguland���nda fet�s �zerinde zararl� etkilere neden olmas� beklenmektedir.
Hayvanlar �zerinde yap�lan �al��malar regorafenibin �reme toksisitesine sahip oldu�unu g�stermi�tir (bkz. B�l�m 5.3).
STIVARGA, kesinlikle gerekli olmad�k�a ve anne i�in sa�layaca�� yararlar� ile fet�s �zerindeki riskleri dikkatle de�erlendirilmeden gebelik d�neminde kullan�lmamal�d�r.
Laktasyon d�nemi
Regorafenib veya metabolitlerinin insan s�t�yle at�l�p at�lmad��� bilinmemektedir. Bununla birlikte s��anlarda, regorafenib veya metabolitleri s�te ge�mektedir.
Emzirilen �ocuk i�in risk g�z ard� edilemez. Regorafenib bebe�in b�y�mesine ve geli�imine zarar verebilir (bkz. B�l�m 5.3).
STIVARGA ile tedavi s�ras�nda emzirme durdurulmal�d�r.
�reme yetene�i/Fertilite
STIVARGA'n�n insan fertilitesi �zerindeki etkisine dair bilgi bulunmamaktad�r. Hayvanlar �zerinde yap�lan �al��malardan elde edilen bulgular, regorafenibin erkek ve di�i fertilitesini azaltabilece�ini g�stermektedir (bkz. B�l�m 5.3).
4.7. Ara� ve makine kullan�m� �zerindeki etkiler
STIVARGA'n�n ara� veya makine kullanma yetene�i �zerindeki etkisine dair herhangi bir �al��ma y�r�t�lmemi�tir. Hastalar�n, STIVARGA tedavisi esnas�nda konsantrasyon ya da reaksiyon yeteneklerini etkileyen belirtiler ya�amas� durumunda, bu etkiler ge�ene kadar ara� ve makine kullanmamalar� tavsiye edilir.
4.8. �stenmeyen etkiler
G�venlilik profilinin �zeti
dahil olmak �zere klinik �al��malarda tedavi edilen 4800'�n �zerinde hastadan elde edilen verilere dayanmaktad�r.
Regorafenibin bu �al��malardaki g�venlilik profili, standart tedavilerin ard�ndan hastal��� progresyon g�steren metastatik kolorektal kanserli 2872 hasta ile y�r�t�len faz III B �al��man�n g�venlilik bulgular� ile uyumludur.
STIVARGA kullanan hastalarda g�zlenen en yayg�n advers ila� reaksiyonlar� (≥%30) a�r�, el-ayak deri reaksiyonu, asteni/yorgunluk, diyare, i�tah kayb� ve besin al�m�n�n azalmas�, hipertansiyon ve enfeksiyondur.
STIVARGA kullanan hastalarda g�zlenen en ciddi advers ila� reaksiyonlar�; �iddetli karaci�er hasar�, hemoraji, gastrointestinal perforasyon ve enfeksiyondur.
Advers reaksiyonlar�n listesi
STIVARGA ile tedavi edilen hastalar ile yap�lan klinik �al��malarda bildirilen advers ila�
reaksiyonlar� a�a��da verilmektedir.
Advers ila� reaksiyonlar�, a�a��da sistem-organ s�n�f� (MedDRA versiyon 14.1) ve s�kl�k derecesine g�re listelenmektedir. Belirli bir reaksiyonu, onun e�anlaml�s�n� ve ili�kili durumlar� tan�mlamak i�in en uygun MedDRA terimi kullan�lm��t�r. �u terimler ve s�kl�k dereceleri kullan�lm��t�r: �ok yayg�n (≥1/10); yayg�n (≥1/100 ila <1/10); yayg�n olmayan (≥1/1000 ila <1/100); seyrek (≥1/10,000 ila <1/1000); �ok seyrek (<1/10,000), bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor).
Her s�kl�k grubu i�inde, istenmeyen etkiler azalan ciddiyet derecesine g�re s�ralanm��t�r.
Enfeksiyonlar ve enfestasyonlar
�ok yayg�n: Enfeksiyon*
�yi huylu ve k�t� huylu neoplazmalar (Kist ve polipler dahil olmak �zere)
Seyrek: Keratoakantoma/skuam�z h�creli deri karsinomu
Kan ve lenf sistemi hastal�klar� �ok yayg�n: Trombositopeni, anemi Yayg�n: L�kopeni
Seyrek: Trombotik mikroanjiyopati (TMA)
�mmun sistem hastal�klar�
Yayg�n olmayan: Hipersensitivite reaksiyonu
Endokrin hastal�klar�
Yayg�n: Hipotiroidizm
Metabolizma ve beslenme hastal�klar�
�ok yayg�n: ��tah kayb� ve besin al�m�n�n azalmas�
Yayg�n: Hipokalemi, hipofosfatemi, hipokalsemi, hiponatremi, hipomagnezemi, hiper�risemi, dehidratasyon
Sinir sistemi hastal�klar�
Seyrek: Geri d�n���ml� posterior l�koensefalopati sendromu
Kardiyak hastal�klar
Yayg�n olmayan: Miyokard infarkt�s�, miyokard iskemisi
Vask�ler hastal�klar
�ok yayg�n: Hemoraji*, hipertansiyon
Yayg�n olmayan: Hipertansif kriz
Bilinmiyor: Anevrizmalar ve arter diseksiyonlar�
Solunum, g���s bozukluklar� ve mediastinal hastal�klar
�ok yayg�n: Disfoni
Gastrointestinal hastal�klar
�ok yayg�n: Diyare, stomatit, kusma, bulant�, kab�zl�k
Yayg�n: Tat alma bozukluklar�, a��z kurulu�u, gastro�zofageal refl�, gastroenterit Yayg�n olmayan: Gastrointestinal perforasyonlar*, gastrointestinal fist�l, pankreatit
Hepato-bilier hastal�klar
�ok yayg�n: Hiperbilirubinemi, transaminazlarda art��
Yayg�n olmayan: �iddetli karaci�er hasar� (karaci�er yetmezli�i dahil)*#
Deri ve deri alt� doku hastal�klar�
�ok yayg�n: El-ayak deri reaksiyonu**, d�k�nt� Yayg�n: Deri kurulu�u, eksfoliyatif d�k�nt�, alopesi
Yayg�n olmayan: T�rnaklarda bozukluk, eritema multiforme Seyrek: Stevens-Johnson sendromu, toksik epidermal nekroliz
Kas-iskelet bozukluklar�, ba� doku ve kemik hastal�klar�
Yayg�n: Kas spazmlar�
B�brek ve idrar yolu hastal�klar�
Yayg�n: Protein�ri
Genel bozukluklar ve uygulama b�lgesine ili�kin hastal�klar
�ok yayg�n: Asteni/yorgunluk, a�r�***, ate�, mukozal inflamasyon
Ara�t�rmalar
�ok yayg�n: Kilo kayb�
Yayg�n: Amilaz art���, lipaz art���, anormal uluslararas� normalizasyon oran� (anormal INR)
�l�mc�l vakalar bildirilmi�tir.
** MedDRA terminolojisinde palmar-plantar eritrodizestezi sendromu.
*** En s�k bildirilen a�r� t�rleri (≥%10) kar�n a�r�s� ve s�rt a�r�s�d�r.
Se�ili advers reaksiyonlar�n tan�m� �iddetli karaci�er hasar�
Klinik �al��malarda fatal sonu�lu �iddetli karaci�er hasar� vakalar� bildirilmi�tir. Bu hastalarda karaci�er fonksiyon bozuklu�u tedavinin ilk 2 ay� i�inde ba�lam�� ve > 20x ULN (Normalin �st S�n�r�) d�zeyinde transaminaz art���n� bilirubindeki art���n takip etti�i hepatosel�ler hasar paterniylekarakterizeolmu�tur.Klinik�al��malarda �l�mc�l sonu�lanan
Hemoraji
Plasebo kontroll� faz III �al��malarda, STIVARGA ile tedavi edilen hastalarda hemorajinin genel insidans� %18,2 ve plasebo alan hastalarda %9,5'tir. STIVARGA ile tedavi edilen hastalarda g�r�len kanama olay� vakalar�n�n �o�u hafif ila orta �iddette olup (Derece 1 ve 2:
%15,2), en belirgin olan� epistaksistir (%6,1). STIVARGA ile tedavi edilen hastalarda �l�mc�l sonu�lar yayg�n olmay�p (%0,7), bu olaylara serebral, solunum ile ilgili, gastrointestinal ve genito-�riner olaylar dahildir.
Enfeksiyon
Plasebo kontroll� faz III �al��malarda, STIVARGA ile tedavi edilen hastalarda plasebo kullanan hastalar ile kar��la�t�r�ld���nda enfeksiyonlar daha yayg�n g�zlenmi�tir (t�m dereceler: %31,6'ya kar�� %17,2). STIVARGA ile tedavi edilen hastalarda en s�k g�r�len enfeksiyonlar hafif ila orta �iddette olup (Derece 1 ve 2: %23,0), idrar yolu enfeksiyonlar� (%5,7), nazofarenjit (4,0%), mukokutan�z ve sistemik mantar enfeksiyonlar�n�n (%3,3) yan� s�ra pn�moni (%2,6) de bunlara dahildir. Enfeksiyonla ili�kili �l�mc�l sonu�lar, STIVARGA ile tedavi edilen hastalarda (%1,0) plasebo alan hastalara (%0,3) k�yasla daha s�k g�zlenmi�tir ve bunlar a��rl�kl� olarak solunum sistemi olaylar�d�r.
El-ayak deri reaksiyonu
Plasebo kontroll� faz III �al��malarda, el ayak deri reaksiyonu (EADR) STIVARGA tedavisi alan hastalarda plasebo alanlara k�yasla daha s�k g�r�lm��t�r (t�m dereceler: %51,4 ve %6,5, KRK; %66,7 ve %15,2 , G�ST ve %51,6 ve %7,3 HSK). STIVARGA ile tedavi edilen hastalarda g�r�len el-ayak deri reaksiyonu vakalar�n�n �o�u tedavinin ilk siklusu s�ras�nda ortaya ��km�� olup, hafif ila orta �iddettedir (Derece 1 ve 2: %34,3, mKRK, %44,7, G�ST ve
% 39,3 HSK). Derece 3 el-ayak deri reaksiyonu insidans� %17,1 (mKRK), %22,0 (G�ST) ve
%12,3 (HSK)'t�r. EADR insidans�n�n STIVARGA tedavisi alan Asyal� hastalarda daha y�ksek oldu�u g�zlenmi�tir (t�m dereceler: %74,8, KRK, %88,2 G�ST ve % 67,1 HSK; 3. Derece: %20,5, KRK, %23,5 G�ST ve %13,5 HSK) (ayr�ca bkz. B�l�m 4.2.).
Hipertansiyon
Plasebo kontroll� faz III �al��malarda, hipertansiyonun genel insidans�, STIVARGA ile tedavi edilen hastalarda, plasebo alanlara k�yasla, daha y�ksektir (% 29,6'ya kar�� % 7,5 KRK, % 60,6'ya kar�� % 25,8 GIST, % 31,0'a kar�� % 6,2 HSK).
STIVARGA ile tedavi edilen hastalarda g�r�len hipertansiyon vakalar�n�n �o�u tedavinin ilk
siklusu s�ras�nda ortaya ��km�� olup, hafif ila orta �iddettedir (Derece 1 ve 2: %20,9, KRK,
% 31,8, G�ST ve %15,8 HSK). Derece 3 hipertansiyon insidans� %8,7 (KRK) % 28,0 (G�ST) ve %15,2 (HSK)'dir. G�ST �al��mas�nda bir kez Derece 4 hipertansiyon vakas� bildirilmi�tir.
Plasebo kontroll� faz III �al��malarda, tedaviye ba�l� protein�rinin genel insidans�, plasebo uygulanan hastalar�n %1,9'una k�yasla STIVARGA ile tedavi edilen hastalarda %9,1 bulunmu�tur.
STIVARGA kolunda bildirilen olaylar�n %35,6's� ile plasebo kolunda bildirilen olaylar�n
%54,5'nn iyile�medi�i/d�zelmedi�i bildirilmi�tir.
T�m klinik �al��malarda, kardiyak bozukluk olaylar� (t�m derecelerde) STIVARGA ile tedavi edilen 75 ya� alt� hastalara (N=4108) k�yasla STIVARGA ile tedavi edilen 75 ya� ve �st� hastalarda (N=410) daha s�k bildirilmi�tir (%6,5 ve %13,7).
Laboratuvar testi anormallikleri
anormallikleri Tablo 3 ve Tablo 4'te yer almaktad�r (ayr�ca bkz. B�l�m 4.4).
Tablo 3: Plasebo kontroll� faz III �al��malarda metastatik CRC (CORRECT), GIST (GRID) ve HSK'l� (RESORCE) hastalarda bildirilen tedaviye ba�l� laboratuvar testi anormallikleri
mCRC (CORRECT) | |||||
Laboratuvar parametreleri | Stivarga art� E�DT (n=500) | Plasebo art� E�DT (n=253) | Stivarga art� E�DT (n=500) | Plasebo art� E�DT (n=253) | |
Derece | |||||
T�m dereceler (%) | 3./4. Derece (%) | ||||
Kan ve lenf sistemi hastal�klar� Hemoglobin say�s�nda azalma Trombositopeni N�tropeni Lenfopeni |
78,5 40,5 2,8 54,1 |
66,3 16,8 0 34,8 |
5,3 2,8 0,6 9,3 |
2,8 0,4 0 4,0 | |
Metabolizma ve beslenme hastal�klar� Hipokalsemi Hipokalemi Hipofasfatemi |
59,3 25,7 57,4 |
18,3 8,3 11,1 |
1,2 4,3 31,1 |
1,2 0,4 3,6 | |
Hepato-biliyer hastal�klar� Hiperbilirubinemi AST art��� ALT art��� |
44,6 65,0 45,2 |
17,1 45,6 29,8 |
12,2 5,9 5,5 |
8,4 5,2 3,2 | |
B�brek ve idrar yolu hastal�klar� Protein�ri |
83,6 |
61,0 |
1,8 |
0,8 | |
Ara�t�rmalar INR art���* Lipaz art��� Amilaz art��� |
23,7 46,0 25,5 |
16,6 18,7 16,7 |
4,2 11,4 2,6 |
1,6 4,4 2,4 | |
GIST (GRID) | |||||
Laboratuvar parametreleri | Stivarga art� E�DT (n=132) | Plasebo art� E�DT (n=66) | Stivarga art� E�DT (n=132) | Plasebo art� E�DT (n=66) | |
Derece | |||||
T�m dereceler (%) | 3./4. Derece (%) | ||||
Kan ve lenf sistemi hastal�klar� Hemoglobin say�s�nda azalma Trombositopeni N�tropeni Lenfopeni |
75,0 12,9 15,9 29,9 |
72,7 1,5 12,1 24,2 |
3,0 0,8 3,1 7,6 |
1,5 1,5 3,0 3,0 | |
Metabolizma ve beslenme hastal�klar� Hipokalsemi Hipokalemi Hipofasfatemi |
16,7 20,5 54,5 |
4,5 3,0 3,1 |
1,5 3,0 21,2 |
0 0 1,5 | |
Hepato-biliyer hastal�klar� Hiperbilirubinemi | 58,3 | 47,0 | 3,8 | 1,5 3,0 | |
AST art��� ALT art��� | 39,4 | 39,4 | 4,6 | 1,5 |
B�brek ve idrar hastal�klar� Protein�ri |
59,2 |
52,5 |
3,1 |
3,4 |
Ara�t�rmalar INR art���* Lipaz art��� Amilaz art��� |
9,3 14,4 - |
12,5 4,6 - |
1,6 0,8 - |
4,7 0 - |
HSK (RESORCE) | ||||
Laboratuvar parametreleri | Stivarga art� E�DT (n=374) | Plasebo art� E�DT (n=193) | Stivarga art� E�DT (n=374) | Plasebo art� E�DT (n=193) |
Derece | ||||
T�m dereceler (%) | 3./4. Derece (%) | |||
Kan ve lenf sistemi hastal�klar� Hemoglobin say�s�nda azalma Trombositopeni N�tropeni Lenfopeni |
72,5 63,1 13,6 67,8 |
71,3 50,0 14,9 58,5 |
6,0 5,4 3,0 17,4 |
4,8 0 1,0 11,7 |
Metabolizma ve beslenme hastal�klar� Hipokalsemi Hipokalemi Hipofasfatemi |
23,4 30,7 70,4 |
10,1 9,0 31,4 |
0,3 4,3 33,9 |
0 2,1 6,9 |
Hepato-biliyer hastal�klar� Hiperbilirubinemi AST art��� ALT art��� |
78,2 92,7 70,4 |
54,5 84,3 58,6 |
15,9 17,8 6,2 |
15,7 19,9 4,7 |
B�brek ve idrar hastal�klar� Protein�ri |
51,0 |
36,5 |
16,7 |
3,1 |
Ara�t�rmalar INR art���* Lipaz art��� Amilaz art��� |
44,4 40,5 23,0 |
35,4 27,0 19,0 |
0,7 14,2 2,8 |
2,1 8,7 2,7 |
4.9. Doz a��m� ve tedavisi
5. FARMAKOLOJ�K �ZELL�KLER
5.1. Farmakodinamik �zellikler
Farmakoterap�tik grup: Di�er antineoplastik ajanlar, protein kinaz inhibit�rleri
ATC kodu: L01EX05
Etki mekanizmas� ve farmakodinamik etkiler:
Regorafenib t�m�r anjiyogenezinde (VEGFR1, -2, -3, TIE2), onkogenezde (KIT, RET, RAF- 1, BRAF, BRAFV600E), metastazda (VEGFR3, PDGFR, FGFR) ve t�m�r imm�nitesinde (CSF1R) yer alan kinazlar dahil olmak �zere �oklu protein kinazlar� g��l� bir �ekilde engelleyen bir oral t�m�r deaktivasyon ilac�d�r. Regorafenib �zellikle gastrointestinal stromal t�m�rlerde maj�r bir onkojenik etken olan mutasyona u�ram�� KIT'i inhibe eder ve
b�ylelikle t�m�r h�cresi proliferasyonunu bloke eder. Klinik �ncesi �al��malarda, regorafenib
kolorektal, gastrointestinal stromal ve hepatosel�ler t�m�r modelleri dahil olmak �zere
bir�ok t�m�r modelinde muhtemel antianjiyogenik ve antiproliferatif etkileri arac�l��� ile g��l� anti-t�m�r aktivite g�stermi�tir. Ek olarak, regorafenib t�m�r ile ili�kili makrofajlar�n seviyelerini d���rm�� ve in vivo anti-metastatik etki g�stermi�tir. Maj�r insan metabolitleri (M-2 ve M-5) in vitro ve in vivo modellerde regorafenib ile kar��la�t�r�ld���nda benzer etkililik sergilemi�lerdir.
Klinik etkililik ve g�venlilik:
Metastatik kolorektal kanser (mKRK)
STIVARGA'n�n klinik etkilili�i ve g�venlili�i, standart tedavi ile ba�ar�s�zl�k sonras�nda progresyon g�stermi� daha �nce yo�un tedavi g�rm�� mKRK'li hastalar ile yap�lan uluslararas�, �ok merkezli, randomize, �ift k�r, plasebo kontroll� bir faz III �al��mas�nda (CORRECT) de�erlendirilmi�tir.
Bu �al��man�n birincil etkililik sonlan�m noktas� genel sa�kal�md�r (OS). �kincil sonlan�m noktalar� progresyonsuz sa�kal�m (PFS), objektif t�m�r yan�t oran� ve hastal�k kontrol oran�d�r.
Toplamda, 760 hasta 2:1 oran�nda 3 hafta boyunca g�nde bir kez oral yolla uygulanan 160 mg regorafenib (her biri 40 mg regorafenib i�eren 4 adet STIVARGA tablet) ile birlikte En �yi Destek Tedavi (E�DT) (N=505) ya da denk plasebo (N=255) ile birlikte E�DT uygulanmas�n� takiben 1 hafta tedavisiz periyoda randomize edilmi�tir. Kullan�lan ortalama g�nl�k regorafenib dozu 147 mg'd�r.
Hastalar, hastal�k progresyonu veya kabul edilemeyen toksisite ortaya ��kana kadar tedaviye devam etmi�tir. 432 �l�m meydana geldi�inde etkililik i�in �nceden planlanm�� bir interim analiz ger�ekle�tirilmi�tir.
OS i�in planlanm�� bu interim analiz, �nceden belirlenmi� etkililik s�n�r�n� ge�tikten sonra, yani plasebo ile birlikte uygulanan E�DT ile kar��la�t�r�ld���nda STIVARGA ile birlikte E�DT uygulanmas�n�n sa�kal�mda uzama sa�lad���n�n kan�t� g�sterildi�inde �al��man�n k�rl��� kald�r�lm��t�r.
Randomize edilen 760 hastan�n medyan ya�� 61 olup, %61'i erkek, %78'i beyaz �rktand�r ve hastalar�n tamam�n�n ba�lang�� ECOG (Eastern Cooperative Oncology Group) performans skoru (PS) 0 veya 1'dir. Hastalar�n %11,4'�nde STIVARGA tedavisi s�ras�nda PS en az 2 bildirilmi�tir.
Doz modifikasyonu ve doz azalt�m� oranlar�n�n yan� s�ra medyan tedavi s�resi ve g�nl�k doz, plasebo al�p PS'nin en az 2 olarak bildirildi�i hastalarda (%8,3) g�zlemlenenle benzerdir. PS'nin en az 2 oldu�u hastalar�n �o�unda tedavi hastal���n progresyonu nedeniyle durdurulmu�tur. PS2 hastalar ve ba�lang��taki dehidratasyonu ≥1 olan hastalar, pivot �al��man�n d���nda tutulmu�tur. Birincil hastal�k b�lgesi kolon (%65), rektum (%29) veya her ikisidir (%6). �al��man�n ba�lang�c�nda hastalar�n %57'sinde KRAS mutasyonu bildirilmi�tir.
Hastalar�n �o�u (%52) metastatik hastal���n tedavisi i�in daha �nce 3 veya daha az basamak tedavi alm��t�r. Bu tedaviler, floropirimidin i�eren kemoterapi, anti-VEGF tedavisi, hasta KRAS do�al tip ise, anti-EGFR tedavisini i�ermi�tir.
STIVARGA'n�n E�DT'ye eklenmesi, plasebonun E�DT'ye eklenmesine k�yasla anlaml� olarak daha uzun sa�kal�m lasonu�lanm��t�r; p de�eri gruplanm�� log-s�ra testine g�re
anlaml� olarak daha uzundur (tehlike oran�: 0,494, p<0,000001, bkz., Tablo 5 ve �ekil 2). Yan�t oran� (tam yan�t veya k�smi yan�t), STIVARGA ve plasebo ile tedavi edilen hastalarda s�ras�yla (p= 0,188432, tek y�nl�) %1 ve %0,4 olmu�tur. Hastal�k kontrol oran� (tam yan�t veya k�smi yan�t ya da stabil hastal�k), STIVARGA ile tedavi edilen hastalarda anlaml� d�zeyde daha y�ksek olmu�tur (%41,0 kar��s�nda %14,9, p<0,000001, tek y�nl�).
Tablo 5: CORRECT �al��mas�ndan elde edilen etkililik sonu�lar�
Etkililik parametresi | Tehlike Oran�* (%95 GA) | P-de�eri (tek tarafl�) | Medyan (%95 GA) | |
STIVARGA + E�DT (N=505) | Plasebo + E�DT (N=255) | |||
Genel Sa�kal�m | 0,774 (0,636, 0,942) | 0,005178 | 6,4 ay (5,9, 7,3) | 5,0 ay (4,4, 5,8) |
Progresyonsuz Sa�kal�m** | 0,494 (0,419, 0,582) | <0,000001 | 1,9 ay (1,9, 2,1) | 1,7 ay (1,7, 1,7) |
* Tehlike oran� <1 STIVARGA lehine.
** Ara�t�rmac�n�n t�m�r yan�t�n� de�erlendirmesine dayan�r.
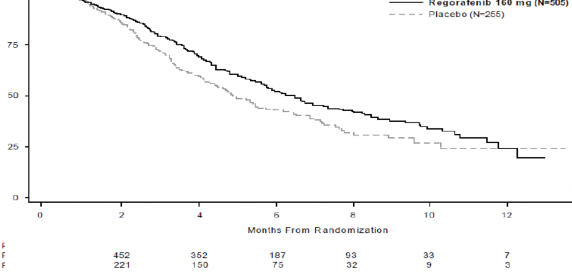
Randomizasyondan sonraki aylar
�ekil 1: Genel Sa�kal�m i�in Kaplan-Meier e�rileri
Genel sa�kal�m ve progresyonsuz sa�kal�m i�in ya� (<65; ≥65), cinsiyet, ECOG PS, primer hastal�k yeri, metastatik hastal���n ilk tan�s�na kadar ge�en s�re, �nceki antikanser tedavisi, metastatik hastal�k i�in �nceki tedavi basamaklar� ve KRAS mutasyon durumuna g�re alt grup analizi, tedavi etkisinin plasebo rejimine k�yasla regorafenib rejimi lehine oldu�unu g�stermi�tir.
�nceki KRAS mutasyon durumuna g�re alt grup analizinin sonu�lar�, KRAS do�al tip t�m�rl� hastalarda GS a��s�ndan plaseboya k�yasla regorafenib lehine tedavi etkisi oldu�unu g�stermektedir. KRAS mutant t�m�rl� hastalarda ise say�ca daha az etki bildirilmi�tir. PFS a��s�ndan KRAS mutasyon tipinden ba��ms�z olarak regorafenib lehine tedavi etkisi g�zlemlenmi�tir. Genel sa�kal�m tehlike oran� (%95 GA), KRAS do�al tip t�m�rl� hastalar i�in 0,653 (0,476 ila 0,895) �eklindeyken KRAS mutant t�m�rl� hastalarda 0,867 (0,670 ila 1,123) bulunmu�tur. Tedavi etkisinde heterojenite kan�t� g�r�lmemi�tir (anlaml� olmayan
etkile�im testi). Progresyonsuz sa�kal�m tehlike oran� (%95 GA), KRAS do�al tip t�m�rl�
hastalar i�in 0,475 (0,362 ila 0,623) �eklindeyken KRAS mutant t�m�rl� hastalarda 0,525
(0,425 ila 0,649) bulunmu�tur
�kinci bir faz III, uluslararas�, �ok merkezli, randomize, �ift k�r, plasebo kontroll� �al��mada (CONCUR) STIVARGA'n�n etkililik ve g�venlili�i �nceden tedavi alm�� metastatik kolorektal kanseri olan ve floropirimidin bazl� kemoterapi ba�ar�s�zl���ndan sonra progresyon g�r�len 204 Asyal� hastada (>%90 Do�u Asyal�) de�erlendirilmi�tir. CONCUR �al��mas�ndaki hastalar�n sadece %59,5'u �ncesinde VEGF veya EGFR hedefli tedaviler ile tedavi edilmi�tir.
Primer etkililik sonlan�m noktas� olarak OS de�erlendirilmi�tir. E�DT'ye STIVARGA eklenmesi plasebo art� E�DT'ye k�yasla anlaml� derecede daha uzun sa�kal�mla sonu�lanm��, tehlike oran�n�n 0,550 (p = 0,000159 gruplanm�� log-s�ra testi) ve medyan GS'nin 8,8 aya k�yasla 6,3 ay oldu�u [%95 GA 0,395, 0,765] belirlenmi�tir.
STIVARGA art� E�DT alan hastalarda PFS'nin de anlaml� derecede daha uzun oldu�u belirlenmi�tir, medyan PFS STIVARGA ile 3,2 ayken plasebo ile 1,7 ayd�r. (tehlike oran�: 0,311, p<0,000001). CONCUR �al��mas�nda STIVARGA art� E�DT'nin g�venlilik profilinin CORRECT �al��mas�nda g�zlenen g�venlilik profiliyle tutarl� oldu�u g�r�lm��t�r.
Gastrointestinal stromal t�m�rler (G�ST)
STIVARGA'n�n klinik etkilili�i ve g�venlili�i, �ncesinde 2 tirozin kinaz inhibit�r� (imatinib ve sunitinib) ile tedavi g�rm�� G�ST'li hastalar ile yap�lan uluslararas�, �ok merkezli, randomize, �ift k�r, plasebo kontroll� bir faz III �al��mas�nda de�erlendirilmi�tir.
Progresyonsuz sa�kal�m (PFS) birincil etkililik sonlan�m noktas� analizi, 144 PFS vakas�ndan sonra ger�ekle�tirilmi�tir (merkezi k�rlenmi� de�erlendirme). Progresyona kadar ge�en s�re (TTP) ve genel sa�kal�mdan (OS) olu�an ikincil sonlan�m noktalar� da de�erlendirilmi�tir (interim analiz).
Toplamda 199 G�ST hastas�, 2:1 oran�nda 3 hafta boyunca ya g�nde bir kez oral yolla uygulanan 160 mg regorafenib ile birlikte E�DT (n=133) ya da denk plasebo ile birlikte E�DT (n=66) uygulanmas�n� takiben 1 hafta tedavisiz periyoda randomize edilmi�tir. Kullan�lan ortalama g�nl�k regorafenib dozu 140 mg'd�r.
Hastalar, hastal�k progresyonu veya kabul edilemeyen toksisite ortaya ��kana kadar tedaviye devam etmi�tir. Plasebo kullanan ve hastal���nda progresyon g�r�len hastalara a��k etiketli regorafenib tedavisi (�apraz ge�i� se�ene�i) olana�� sunulmu�tur. Regorafenib kullanan ve hastal���nda progresyon g�r�len ve ara�t�rmac�n�n kanaatine g�re regorafenib ile tedavinin klinik fayda sa�lamakta oldu�u hastalara a��k etiketli regorafenibe devam etme imkan� verilmi�tir.
Randomize edilen 199 hastan�n ortalama ya�� 58 olup %64'� erkek, %68'i beyaz �rktand�r ve hastalar�n tamam�n�n ba�lang�� ECOG Performans Skoru 0 veya 1'dir. En yak�n progresyon veya relapstan bu yana ge�en genel medyan s�re 6 hafta olarak belirlenmi�tir.
E�DT ile regorafenib kullan�lmas�, plasebo ile birlikte uygulanan E�DT'ye k�yasla anlaml� derecede daha uzun PFS ile sonu�lanm�� olup, tehlike oran� 0,268 [%95 GA 0,185, 0,388] ve medyan PFS 0,9 aya kar��n 4,8 ayd�r (p<0,000001). Hastal�k progresyonu veya �l�m i�in
ba��l risk regorafenib ile tedaviedilenhastalarda, plasebo ile tedavi edilen hastalar ile
ya�, cinsiyet, co�rafik b�lge, �nceki tedavi basamaklar� ve ECOG performans skorundan ba��ms�z olarak tutarl� olmu�tur.
TTP, E�DT ile birlikte regorafenib kullanan hastalarda, E�DT ile birlikte plasebo uygulanan hastalara k�yasla anlaml� derecede daha uzun olmu� olup, tehlike oran� 0,248 [%95 GA 0,170, 0,364] ve medyan TTP 0,9 aya kar��n 5,4 ayd�r (p<0,000001) (bkz., Tablo 6).
Ba�lang��ta plasebo koluna randomize edilmi� hastalar�n %85'inde progresyon sonras� �apraz ge�i� ger�ekle�mi� olmakla birlikte, OS analizinin tehlike oran�, pozitif tedavi etkisi y�n�nde e�ilim g�stermi�tir (tehlike oran�=0,772 [%95 GA, 0,423, 1,408]; p=0,199; iki kolda da medyan OS'ye ula��lamam��t�r) (bkz., Tablo 6, �ekil 4).
Tablo 6 : GRID �al��mas�ndan elde edilen etkililik sonu�lar�
Etkililik | Tehlike Oran�* | P-de�eri | Medyan (%95 GA) | |
parametresi | (%95 GA) | (tek tarafl�) | STIVARGA + E�DT (N=133) | Plasebo + E�DT (N=66) |
Progresyonsuz Sa�kal�m | 0,268 (0,185, 0,388) | <0,000001 | 4,8 ay (4,0, 5,7) | 0,9 ay (0,9, 1,1) |
Progresyona Kadar Ge�en S�re | 0,248 (0,170, 0,364) | <0,000001 | 5,4 ay (4,1, 5,7) | 0,9 ay (0,9, 1,1) |
Genel Sa�kal�m | 0,772 (0,423, 1,408) | 0,199 | Ula��lamam��t�r | Ula��lamam��t�r |
GA: G�ven aral���
* Tehlike oran� <1 STIVARGA lehine.
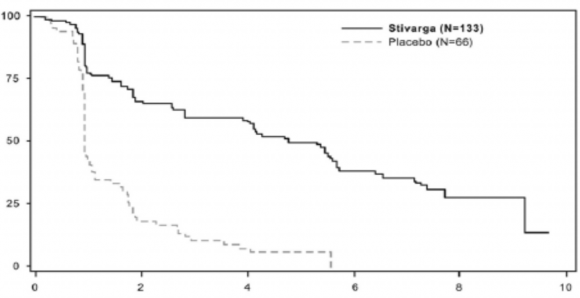
�ekil 2: Progresyonsuz Sa�kal�m i�in Kaplan-Meier e�rileri
Progresyonsuz Sa�kal�m Olas�l��� (%)
Randomizasyondan sonraki aylar
![]()
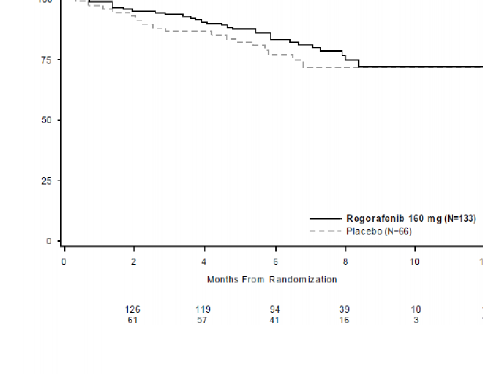
Genel Sa�kal�m Olas�l��� (%)
�ekil 3: Genel sa�kal�m i�in Kaplan-Meier e�rileri
Ek olarak, E�DT ile plasebo uygulanan 56 hasta, hastal�k progresyonunu takiben �apraz ge�i�ten sonra a��k etiketli STIVARGA kullanm��t�r ve E�DT ile STIVARGA uygulanan toplam 41 hasta, hastal�k progresyonunu takiben STIVARGA tedavisine devam etmi�tir. Medyan ikincil PFS (ara�t�rmac� de�erlendirmelerine g�re �l��ld���nde) s�ras�yla 5,0 ve 4,5 ayd�r.
Hepatosel�ler karsinom (HSK)
STIVARGA'n�n klinik etkililik ve g�venlili�i, daha �nce sorafenib tedavisi alm�� hepatosel�ler karsinomlu hastalarla y�r�t�len uluslararas�, �ok merkezli, randomize, �ift k�r, plasebo kontroll� bir faz III �al��mada (RESORCE) de�erlendirilmi�tir.
Primer etkililik sonlan�m noktas� Genel Sa�kal�m (GS) olmu�tur. Sekonder sonlan�m noktalar� Progresyonsuz Sa�kal�m (PFS), Progresyona Kadar Ge�en S�re (TTP), Objektif T�m�r Yan�t Oran� (ORR) ve Hastal�k Kontrol Oran�d�r (DCR).
Demografik �zellikler ve ba�lang�� �zellikleri STIVARGA ve plasebo tedavisi alan gruplarda
benzerdir ve randomize edilen 573 hasta i�in a�a��daki tabloda g�sterilmi�tir:
Medyan ya�: 63 ya�
5.2. Farmakokinetik �zellikler
Genel �zelliklerEmilim:
Her biri 40 mg i�eren 4 tablet olarak verilen 160 mg'l�k tek bir oral regorafenib dozundan yakla��k 3 ila 4 saat sonra, regorafenib yakla��k 2,5 mg/L'lik ortalama doruk plazma d�zeyine ula��r. Tabletlerin ortalama ba��l biyoyararlan�m� oral ��zeltiye k�yasla %69-83't�r. Regorafenib ve farmakolojik olarak aktif maj�r metabolitlerinin M-2 (N-oksit) ve M-5 (N- oksit ve N-desmetil) konsantrasyonlar�, ilac�n az ya�l� (hafif) bir kahvalt�n�n ard�ndan verilmesi durumunda ya�dan zengin bir kahvalt� sonras�nda ya da a�l�k ko�ullar�nda verilmesine k�yasla en y�ksektir. Regorafenibe maruziyet a�l�k ko�ullar�na k�yasla, ilac�n ya�dan zengin bir kahvalt� ile verilmesi durumunda %48, az ya�l� bir kahvalt� ile verilmesi durumunda ise %36 artm��t�r. M-2 ve M-5 metabolitlerine maruziyet, regorafenibin az ya�l� bir kahvalt� ile verilmesi durumunda a�l�k durumuna k�yasla daha y�ksek, ya�dan zengin bir yemek ile verilmesi durumunda ise a�l�k ko�ullar�nda verilmesine k�yasla daha d���k olmu�tur.
Da��l�m:
Regorafenibin yan� s�ra dola��mdaki maj�r metabolitler i�in plazma konsantrasyon-zaman profilleri, 24 saatlik doz uygulama aral���nda enterohepatik dola��ma ba�l� olarak �ok say�da pik g�stermi�tir.
Regorafenibin insan plazma proteinlerine in vitro protein ba�lanmas� y�ksektir (%99,5). Biyotransformasyon:
Regorafenib esas olarak karaci�erde metabolize olur, CYP3A4'�n arac�l�k etti�i oksidatif metabolizmaya girerken, ayn� zamanda UGT1A9 arac�l���yla glukuronidasyona da u�rar. Plazmada iki maj�r ve alt� min�r regorafenib metaboliti tan�mlanm��t�r. Regorafenibin plazmada dola�an esas metabolitleri, farmakolojik olarak aktif olan ve kararl� durumda regorafenib ile benzer konsantrasyonlara sahip M-2 (N-oksit) ve M-5 (N-oksit ve N- desmetil)'tir.
M-2 ve M-5'in in vitro proteine ba�lanmalar� regorafenibe g�re daha y�ksektir (s�ras�yla
%99,8 ve %99,95).
Metabolitleri, gastrointestinal sistemde mikrobiyal flora taraf�ndan indirgenebilir veya hidrolize olabilir ve bu da konjuge olmayan ila� ve metabolitlerin geri emilmelerini sa�layabilir (enterohepatik dola��m).
Eliminasyon:
Oral uygulamay� takiben, plazmadaki regorafenib ve metabolit M-2 i�in ortalama eliminasyon yar�lanma �mr� farkl� �al��malarda 20 ila 30 saat aras�nda de�i�mektedir. Metabolit M-5 i�in ortalama eliminasyon yar�lanma �mr� yakla��k 60 saattir (40 ila 100 saat aras�nda).
Radyoaktif dozun yakla��k %90'� uygulamay� takiben 12 g�n i�inde geri kazan�lm�� ve dozun yakla��k %71'i fe�es ile (%47 ana bile�en, %24 metabolit olarak) ve dozun yakla��k %19'u idrarda glukuronize metabolitler �eklinde at�lm��t�r. Kararl� durum ko�ullar�nda glukuronidlerin �riner itrah� %10'un alt�na d��m��t�r. Fe�este bulunan ana bile�ik, glukuronidlerin intestinal degradasyonundan veya M-2 metabolitinin (N-oksit) ve emilime u�ramam�� regorafenibin red�ksiyonundan elde edilebilmi�tir. Gastrointestinal kanalda mikrobiyal flora M-5 metabolitini M-4'e indirgeyerek M-4 i�in geri emilime (enterohepatik dola��m) imkan verebilir. M-5 sonu� olarak fe�este M-4 �zerinden M-6 �eklinde at�lmaktad�r.
Do�rusall�k/Do�rusal olmayan durum:
Kararl� durumda regorafenibin sistemik maruziyeti 60 mg'a kadar dozla orant�l� olarak ve
60 mg'dan daha y�ksek dozlarda dozla orant�l� de�erden daha d���k bir de�erde art�� g�stermektedir. Kararl� durumda regorafenib birikimi plazma konsantrasyonlar�nda yakla��k 2 katl�k bir art��la sonu�lan�r ve bu, eliminasyon yar�lanma �mr� ve doz uygulama s�kl��� ile tutarl�d�r. Kararl� durumda, 160 mg regorafenibin oral uygulamas�ndan sonra regorafenib yakla��k 3,9 mg/L'lik (8,1 mikromolar) ortalama doruk plazma d�zeylerine eri�ir ve ortalama plazma konsantrasyonlar�n�n tepe:vadi oran� 2'den d���kt�r.
Her iki metabolit de (M-2 ve M-5) do�rusal olmayan birikim g�stermektedir. Tek doz regorafenibin verilmesinden sonra M-2 ve M-5'in plazma konsantrasyonlar�, ana bile�ene oranla �ok daha d���k olmakla birlikte, kararl� durumda plazma konsantrasyonlar� regorafenib ile benzerdir.
Hastalardaki karakteristik �zellikler
B�brek yetmezli�i olan hastalar:
Mevcut klinik verilere ve fizyoloji bazl� farmakokinetik modellemeye g�re, regorafenibin ve metabolitleri M-2 ve M-5'in kararl� durum maruziyeti hafif ve orta derecede b�brek yetmezli�i olan hastalar ve b�brek fonksiyonlar� normal olan hastalarda benzerdir.
�iddetli b�brek yetmezli�i olan hastalar ile normal b�brek fonksiyonu olan hastalar kar��la�t�r�ld���nda, regorafenib maruziyeti benzerken, klinik olarak anlaml� kabul edilmeyen kararl� durum ko�ullar�nda M-2 ve M-5 maruziyeti yakla��k %30 oran�nda azalm��t�r.
Regorafenibin farmakokineti�i son evre b�brek yetersizli�i olan hastalarda ara�t�r�lmam��t�r. Bununla birlikte, fizyoloji bazl� farmakokinetik modelleme, bu hastalarda maruziyette g�r�lebilecek herhangi bir anlaml� de�i�ikli�i �ng�rmemektedir.
Karaci�er yetmezli�i olan hastalar:
Regorafenibin ve metabolitleri olan M-2 ve M-5'in maruziyeti, hafif derecede karaci�er yetmezli�i olan (Child-Pugh A) hastalarda ve karaci�er fonksiyonlar� normal olan hastalarda benzerdir. 100 mg tek doz regorafenib uygulanmas�ndan sonra orta derecede karaci�er yetmezli�i (Child-Pugh B) olan hastalardaki maruziyetinin, karaci�er fonksiyonlar� normal olan hastalardaki farmakokineti�i ile benzer oldu�una ili�kin s�n�rl� veri bulunmaktad�r. Child-Pugh C (�iddetli) karaci�er yetmezli�i olan hastalara ili�kin bilgi bulunmamaktad�r. Regorafenib ba�l�ca karaci�er yoluyla at�lmaktad�r ve bu hasta pop�lasyonunda maruziyet artabilmektedir.
Geriyatrik hastalar:
Regorafenibin farmakokineti�i, �al���lan ya� aral���nda (29 – 85 ya�), ya�tan etkilenmemi�tir.
Cinsiyet:
Regorafenibin farmakokineti�i cinsiyetten etkilenmemektedir.
Etnik farkl�l�klar:
Farkl� Asya pop�lasyonlar�ndaki (�inli, Japon, Koreli) regorafenib maruziyeti beyaz �rkta g�r�len ile ayn� aral�ktad�r.
Kardiyak Elektrofizyoloji/QT uzamas�:
Erkek ve kad�n kanser hastalar�nda yap�lan �zel bir QT �al��mas�nda, kararl� durumda 160
5.3. Klinik �ncesi g�venlilik verileri
Sistemik toksisite
Farelere, s��anlara ve k�peklere tekrarlanan doz uygulanmas�ndan sonra esas olarak b�breklerde, karaci�erde, sindirim sisteminde, kalpte, tiroid bezinde, kan ve lenf sisteminde, endokrin sistemde, �reme sisteminde ve deride olmak �zere bir dizi organda advers etkiler g�zlenmi�tir. S��anlarda 26 haftal�k tekrar dozlu toksisite �al��mas�nda kalpte atrioventrik�ler damarlarda kal�nla�ma insidans�nda hafif art�� g�r�lm��t�r. Nedeni ya�a ba�l� fizyolojik proseslerin h�zlanmas� olabilir. Bu etkiler, �ng�r�len insan maruziyeti aral���nda veya alt�ndaki sistemik maruziyetlerde meydana gelmi�tir (EAA kar��la�t�rmas�na dayal� olarak).
Gen� ve b�y�me d�nemindeki hayvanlar�n ve yavru s��anlar�n di�lerinde ve kemiklerinde de�i�iklikler ve �reme sistemindeki advers etkiler daha belirgin olup, bu durum �ocuklar ve ergenler i�in potansiyel bir riske i�aret etmektedir.
Genotoksisite ve karsinojenisite
Regorafenibin karsinojenik potansiyeline ili�kin �al��ma y�r�t�lmemi�tir.
Fareler �zerinde y�r�t�len in vitro ve in vivo standart analizlerde, regorafenibin genotoksik potansiyeline ili�kin bulgu g�zlenmemi�tir.
�reme ve geli�im toksisitesi
Fertiliteye �zg� �al��malar y�r�t�lmemi�tir. Bununla birlikte, s��anlarda ve k�peklerde �ng�r�len insan maruziyeti alt�ndaki maruziyetlerde (EAA kar��la�t�rmas�na dayal� olarak) tekrarlanan doz uygulamas�n� takiben testislerde, overlerde ve uterusta g�zlenen morfolojik de�i�ikliklere dayal� olarak regorafenibin erkek ve di�i �reme sistemini advers olarak etkileme potansiyelinin oldu�u dikkate al�nmal�d�r. G�zlenen de�i�iklikler sadece k�smen geri d�n���ml�d�r.
Tav�anlarda, �ng�r�len insan maruziyetinin alt�ndaki maruziyetlerde (EAA kar��la�t�rmas�na dayal� olarak) regorafenibin intrauterin geli�im �zerindeki etkisi g�sterilmi�tir. Ba�l�ca bulgular �riner sistem, kalp ve maj�r damarlar ve iskelette malformasyonlar� i�ermi�tir.
�evresel Risk De�erlendirme (�RD)
�evresel risk de�erlendirme �al��malar�, regorafenibin �evreye kar�� kal�c�, biyoak�m�latif ve toksik olma potansiyeline sahip oldu�unu; yer�st� suyu ve ��kelti k�s�mlar�na risk olu�turabildi�ini g�stermi�tir.
6. FARMAS�T�K �ZELL�KLER
6.1. Yard�mc� maddelerin listesi
Mikrokristalin sel�loz Kroskarmelloz sodyum Magnezyum stearatPovidon
Silika, kolloidal susuz K�rm�z� demir oksit – E172 Sar� demir oksit – E172 Lesitin (soyadan elde edilir) Makrogol
Polivinil alkol, k�smen hidrolize
Talk
Titanyum dioksit – E171
6.2. Ge�imsizlikler
Bilinen herhangi bir ge�imsizli�i bulunmamaktad�r.
6.3. Raf �mr�
36 ay
�i�e ilk a��ld�ktan sonra s�k�ca kapat�lmal�d�r. �i�e bir kez a��ld�ktan sonra t�bbi �r�n�n nem tutucu yoklu�unda dahi 7 hafta (49 g�n) stabil oldu�u g�sterilmi�tir. Ard�ndan t�bbi �r�n imha edilmelidir.
6.4. Saklamaya y�nelik �zel tedbirler
30°C'nin alt�ndaki oda s�cakl���nda saklay�n�z. Nemden korumak i�in orijinal ambalaj�nda saklay�n�z. Nem tutucu kaps�l� �i�eden ��karmay�n�z.
6.5. Ambalaj�n niteli�i ve i�eri�i
STIVARGA karton kutulara yerle�tirilen ve her birinde 28 tablet ve 1 adet nem tutucu kaps�l i�eren �ocuk emniyetli, s�zd�rmaz polipropilen kapakl�, beyaz opak 3 adet HDPE �i�ede ambalajlanmaktad�r (bir kutuda toplam 28 tablet x 3 �i�e = 84 tablet).
6.6. Be�eri t�bbi �r�nden arta kalan maddelerin imhas� ve di�er �zel �nlemler
�zerindeki talimatlara uygun �ekilde kapa�� bast�rarak sola do�ru �eviriniz. �i�e ilk a��ld�ktan sonra s�k�ca kapat�lmal�d�r. Nem tutucu kaps�l yutulmamal�d�r.
Bu t�bbi �r�n, �evreye kar�� risk olu�turabilmektedir (bkz. B�l�m 5.3).
Regorafenibin kullan�m�, yer�st� suyu ve ��kelti k�s�mlar�nda risk ile sonu�lanabilmektedir.
Bu nedenle STIVARGA, at�k sular (kanalizasyon) veya ev at�klar� ile birlikte at�lmamal�d�r.
Kullan�lmam�� olan �r�nler ya da at�k materyaller “T�bbi At�klar�n Kontrol� Y�netmeli�i” ve “Ambalaj At�klar�n�n Kontrol� Y�netmelik”lerine uygun olarak imha edilmelidir.
 Y�ksek Tansiyon
Hipertansiyon s�rekli anormal derecede y�ksek olan kan bas�nc�d�r. Tansiyon
atardamarlar�n�zdaki kan�n bas�nc�d�r.
Y�ksek Tansiyon
Hipertansiyon s�rekli anormal derecede y�ksek olan kan bas�nc�d�r. Tansiyon
atardamarlar�n�zdaki kan�n bas�nc�d�r. |
 Rahim Boyu ( Serviks ) Kanseri
Rahim boynu (serviks) kanseri 35 ya� alt� kad�nlarda g�r�len vakalarda meme kanserinden
sonra ikinci s�ray� al�r.Serviks kanserinin geli�mesi y�llarca s�rebilir.
Rahim Boyu ( Serviks ) Kanseri
Rahim boynu (serviks) kanseri 35 ya� alt� kad�nlarda g�r�len vakalarda meme kanserinden
sonra ikinci s�ray� al�r.Serviks kanserinin geli�mesi y�llarca s�rebilir. |
 |
Belso�uklu�u, Chlamydia ve Frengi Belso�uklu�u, bakterilerin sebep oldu�u bir enfeksiyondur. Cinsel ili�ki yoluyla bula��r ve d�lyata�� boynunda, idrar yollar�nda, an�ste, makatta ve bo�azda enfeksyona sebep olabilir. |
 |
A��r� Alkol Kullan�m�, Alkolizm Alkol ba��ml�l���, alkol kullan�m� ve alkol sorunlar� aras�ndaki fark� a��klamak g��t�r. �rne�in, ge�mi�te alkol kullanm�� olan bir kimsenin mutlaka alkol ba��ml�s� olmas� gerekmez. |
 |
S�rt A�r�s� S�rt a�r�s� birden bire ortaya ��k�p �iddetli (akut) olabilir veya zamanla geli�ip daha uzun s�reli sorunlara (kronik) neden olabilir. |
�LA� GENEL B�LG�LER�
Bayer T�rk Kimya San. Tic. Ltd. �ti.
| Geri �deme Kodu | A15618 |
| Sat�� Fiyat� | 73173.5 TL [ 9 Jun 2026 ] |
| �nceki Sat�� Fiyat� | 73173.5 TL [ 22 May 2026 ] |
| Original / Jenerik | Original �la� |
| Re�ete Durumu | Normal Re�eteli bir ila�d�r. |
| Barkodu | 8699546090303 |
| Etkin Madde | Regorafenib |
| ATC Kodu | L01EX05 |
| Birim Miktar | 40 |
| Birim Cinsi | MG |
| Ambalaj Miktar� | 84 |
| Antineoplastik ve �mm�nomod�lat�r Ajanlar |
| �thal ( ref. �lke : Fransa ) ve Be�eri bir ila�d�r. |