ZEJULA 100 mg film kapl� tablet (56 adet) K�sa �r�n Bilgisi
{ Niraparib }
1. BE�ER� TIBB� �R�N�N ADI
ZEJULA 100 mg film kapl� tablet Sitotoksik
2. KAL�TAT�F VE KANT�TAT�F B�LE��M
Etkin madde
Her bir film kapl� tablet 100 mg niraparibe e�de�er 159,3 mg niraparib tosilat monohidrat
i�ermektedir.
Yard�mc� maddeler
Her bir film kapl� tablet 34,66 mg laktoz monohidrat (s���r s�t�nden elde edilir) i�ermektedir.
Yard�mc� maddelerin tam listesi i�in B�l�m 6.1'e bak�n�z.
3. FARMAS�T�K FORMU
Film kapl� tablet (tablet).
Bir taraf�nda “100”, di�er taraf�nda “Zejula” bask�s� bulunan gri renkli, oval �eklinde film kapl�
tablet.
/4. derece trombositopeninin ba�lang�c�na kadar ge�en medyan s�re s�ras�yla 22 ve 23 g�nd�r.
4. KL�N�K �ZELL�KLER
4.1. Terap�tik endikasyonlar
ZEJULA
Birinci basamak over kanseri idame tedavisinde
FIGO evre 3 veya 4, y�ksek gradl� ser�z epitelyal over, fallop t�p� veya primer peritoneal kanser hastas� olan ve birinci basamak platin temelli kemoterapinin tamamlanmas�ndan sonra yan�t veren (tam veya k�smi), ECOG performans durumu 0-1 olan yeti�kin hastalarda idame tedavisi i�in monoterapi olarak endikedir. Tedavi s�resi hastal�k ilerlemesine veya tolere edilemeyen toksisiteye kadar veya maksimum 3 y�ld�r.
Rek�rren (Tekrarlayan) over kanseri idame tedavisinde
Platinli tedavinin tamamlanmas�ndan en az 6 ay veya sonras�nda n�ks eden platin duyarl�, y�ksek gradl� ser�z epitelyal over, fallop t�p� veya primer peritoneal kanser hastas� olan ve n�ks nedeniyle uygulanan platin esasl� kemoterapiye yan�t veren (tam veya k�smi), ECOG performans durumu 0-1 olan yeti�kin hastalarda idame tedavisi i�in monoterapi olarak kullan�mda endikedir.
4.2. Pozoloji ve uygulama �ekli
ZEJULA tedavisi, kanser tedavi �r�nlerinin kullan�m�nda deneyimli bir hekim taraf�ndan ba�lat�lmal� ve bu hekimin g�zetiminde y�r�t�lmelidir.
Pozoloji
Birinci basamak over kanseri idame tedavisi
�nerilen ZEJULA ba�lang�� dozu g�nde bir kez 200 mg'd�r (iki adet 100 mg tablet). Bununla birlikte 77 kg veya daha fazla kiloda olan ve bazal trombosit say�m� 150.000 mcgLve daha fazla olan hastalarda, �nerilen ZEJULA ba�lang�� dozu g�nde bir kez 300 mg'd�r (�� adet 100 mg tablet) (bkz. B�l�m 4.4 ve 4.8).
Rek�rren (Tekrarlayan) over kanseri idame tedavisi
Doz, toplam g�nl�k doz olarak 300 mg'a e�de�er, g�nde bir kez �� adet 100 mg tablettir.
Hastalar dozlar�n� her g�n yakla��k ayn� saatte almaya te�vik edilmelidir. Uyku �ncesinde kullan�m, bulant�n�n y�netilmesinde potansiyel bir y�ntem olabilir.
Hastal�kta ilerleme veya toksisite olana kadar tedaviye devam edilmesi �nerilmektedir.
Dozun ka��r�lmas�
Hasta bir dozu ka��r�rsa, sonraki dozu d�zenli olarak planlanm�� saatinde almal�d�r.
Advers reaksiyonlar i�in doz ayarlamas�
Advers reaksiyonlar i�in �nerilen doz de�i�iklikleri Tablo 1, 2 ve 3'te listelenmektedir.
Genel olarak, advers reaksiyonun ge�mesi i�in hastalara zaman vermek amac�yla tedaviye ara verilmesi (ard���k 28 g�nden fazla olmamak kayd�yla) ve daha sonra tedaviye ayn� dozda devam edilmesi �nerilmektedir. Advers reaksiyonun tekrar ger�ekle�mesi durumunda, tedaviye ara verilmesi ve daha d���k bir dozda devam edilmesi �nerilmektedir. 28 g�nl�k aradan sonra da advers reaksiyonlar�n devam etmesi durumunda, ZEJULA'ya devam edilmemesi �nerilmektedir. E�er advers reaksiyonlar, dozlara ara verilmesi ya da dozun azalt�lmas� gibi y�ntemlerle y�netilemiyorsa, ZEJULA kullan�m�n�n kesilmesi �nerilmektedir.
Tablo 1: Advers reaksiyonlar i�in �nerilen doz de�i�iklikleri | ||
Ba�lang�� doz d�zeyi | 200 mg | 300 mg |
Birinci doz azaltma | 100 mg/g�n | 200 mg/g�n (iki adet 100 mg tablet) |
�kinci doz azaltma | �la� kullan�m� kesilmelidir. | 100 mg/g�n* (bir adet 100 mg tablet) |
Dozun 100 mg/g�n'�n alt�na azalt�lmas� gerekiyorsa, ZEJULA kullan�m� kesilmelidir.
Tablo 2: Hematolojik olmayan advers reaksiyonlar i�in doz modifikasyonu
Profilaksisin uygulanamayaca��n�n d���n�ld��� veya advers reaksiyonun tedaviye kar��n devam etti�i, hematolojik olmayan CTCAE* ≥ 3. derece tedaviyle ili�kili advers reaksiyon | �lk kez ger�ekle�ti�inde: |
�kinci kez ger�ekle�ti�inde: | |
Hastaya ZEJULA 100 mg/g�n verilirken 28 g�nden uzun s�ren CTCAE ≥ 3. derece tedaviyle ili�kili advers reaksiyon | Tedavi sonland�r�lmal�d�r. |
ZEJULA'ya maksimum 28 g�n boyunca olmak kayd�yla veya advers reaksiyon sona erinceye ya da ortadan kalk�ncaya kadar ara verilmesi.
Tablo 1'de belirtildi�i gibi, ZEJULA'n�n daha d���k bir doz d�zeyinde devam ettirilmesi.
ZEJULA'ya maksimum 28 g�n boyunca olmak kayd�yla veya advers reaksiyon sona erinceye ya da ortadan kalk�ncaya kadar ara verilmesi.
Tablo 1'de belirtildi�i gibi, ZEJULA'ya daha d���k bir doz d�zeyinde devam ettirilmesi veya kullan�m�n�n kesilmesi.
4.3. Kontrendikasyonlar
Etkin madde
Emzirme (bkz. B�l�m 4.6).
4.4. �zel kullan�m uyar�lar� ve �nlemleri
Hematolojik advers reaksiyonlar
ZEJULA ile tedavi edilen hastalarda hematolojik advers reaksiyonlar (trombositopeni, anemi, n�tropeni) bildirilmi�tir (bkz. B�l�m 4.8). V�cut a��rl��� veya bazal trombosit say�m� d���k olan hastalarda 3. derece ve �zeri trombositopeni riski daha y�ksek olabilir (bkz. B�l�m 4.2).
Tedavi s�ras�nda t�m hematolojik parametrelerin herhangi birindeki klinik a��dan anlaml� de�i�imleri izlemek i�in, ilk ay boyunca haftal�k tam kan say�m�, ard�ndan izleyen 10 ayl�k tedavi boyunca ayl�k izlem ve bu s�reden sonra da periyodik izlem �nerilmektedir (bkz. B�l�m 4.2).
Hasta, ara verildikten sonraki 28 g�n i�inde ge�meyen pansitopeni dahil olmak �zere �iddetli persistan hematolojik toksisite geli�tirirse, ZEJULA kullan�m� kesilmelidir.
Trombositopeni riski nedeniyle, antikoag�lanlar ve trombosit say�m�n� d���rd��� bilinen t�bbi �r�nler dikkatli kullan�lmal�d�r (bkz. B�l�m 4.8).
Miyelodisplastik sendrom/akut miyeloid l�semi
Klinik �al��malarda ve pazara verilme sonras�nda ZEJULA monoterapisi veya kombinasyon tedavisiyle tedavi edilen hastalarda miyelodisplastik sendrom/akut miyeloid l�semi (MDS/AML) olgular�, �l�mle sonu�lanan vakalar dahil, g�zlemlenmi�tir (bkz. B�l�m 4.8).
Klinik �al��malarda hastalarda MDS/AML geli�meden �nceki ZEJULA tedavisi s�resi 0,5 ay ila >4,9 y�l aral���ndad�r. Olgular sekonder, kanser tedavisiyle ili�kili MDS/AML a��s�ndan tipiktir. T�m hastalara platin i�eren kemoterapi rejimi uygulanm�� ve bir�o�una ayn� zamanda DNA'ya zarar veren ba�ka ajanlar ve radyoterapi uygulanm��t�r. Baz� hastalarda kemik ili�i
bask�lanmas� �yk�s� vard�r. NOVA �al��mas�nda, MDS/AML insidans� gBRCAmut kohortunda (%7,4), gBRCAmut olmayan kohorta (%1,7) g�re daha y�ksektir.
��pheli MDS/AML veya uzun s�reli hematolojik toksisiteler i�in, hasta ileri de�erlendirme i�in bir hematolo�a sevk edilmelidir. MDS/AML tan�s� do�rulan�rsa, ZEJULA tedavisi kesilmeli ve hasta uygun �ekilde tedavi edilmelidir.
Hipertansif kriz dahil olmak �zere hipertansiyon
ZEJULA kullan�m� s�ras�nda hipertansif kriz dahil olmak �zere hipertansiyon bildirilmi�tir (bkz. B�l�m 4.8). ZEJULA tedavisine ba�lamadan �nce, var olan hipertansiyon yeterli d�zeyde kontrol alt�na al�nmal�d�r. �ki ay boyunca en az haftada bir kez, ard�ndan ilk y�l boyunca ayda bir kez, daha sonra ZEJULA tedavisi s�ras�nda periyodik olarak kan bas�nc� izlemi yap�lmal�d�r. Uygun hastalarda kan bas�nc�nda art�� olmas� durumunda doktorlar�na ba�vurma talimat� verilerek evde kan bas�nc� izlemi d���n�lebilir.
Hipertansiyon gerekirse hem antihipertansif t�bbi �r�nlerle hem de ZEJULA dozunun ayarlanmas�yla t�bbi olarak y�netilmelidir (bkz. B�l�m 4.2). Klinik programda, hastalar ZEJULA tedavisi g�r�rken her bir 28 g�nl�k d�ng�n�n 1. g�n�nde kan bas�nc� �l��m� yap�lm��t�r. �o�u olguda, hipertansiyon ZEJULA dozunda ayarlama yap�larak veya yap�lmaks�z�n standart antihipertansif tedavi kullan�larak yeterli d�zeyde kontrol edilmi�tir (bkz. B�l�m 4.2). Hipertansif kriz durumunda veya t�bbi a��dan anlaml� hipertansiyonun antihipertansif tedaviyle yeterli d�zeyde kontrol edilemedi�i durumlarda ZEJULA kullan�m� kesilmelidir.
Posterior Reversibl Ensefalopati Sendromu (PRES)
ZEJULA alan hastalarda Posterior Reversibl Ensefalopati Sendromu (PRES) raporlar� bildirilmi�tir (bkz. B�l�m 4.8). PRES, seyrek g�r�len, geri d�n��l� (reversibl) bir n�rolojik hastal�kt�r ve ili�kili hipertansiyon varl���nda veya yoklu�unda n�bet, ba� a�r�s�, zihinsel durum de�i�ikli�i, g�rme bozuklu�u veya kortikal k�rl�k dahil olmak �zere h�zla de�i�en semptomlarla ortaya ��kabilir. PRES tan�s� konmas� i�in, tercihen manyetik rezonans g�r�nt�lemesiyle (MRI) beyin g�r�nt�leme yap�larak do�rulanmas� gerekmektedir.
PRES olgular�nda, ZEJULA kullan�m�n�n kesilmesi ve hipertansiyon dahil olmak �zere spesifik semptomlar�n tedavi edilmesi �nerilmektedir. Ge�mi�te PRES deneyimleyen hastalarda ZEJULA tedavisinin tekrar ba�lat�lmas�n�n g�venlili�i bilinmemektedir.
Gebelik/kontrasepsiyon
ZEJULA gebelik d�neminde veya �ocuk do�urma potansiyeli olan ancak tedavi s�ras�nda ve son ZEJULA dozundan sonra 6 ay boyunca y�ksek etkili kontrasepsiyon kullanmak istemeyen kad�nlarda kullan�lmamal�d�r (bkz. B�l�m 4.6). Tedaviden �nce �ocuk do�urma potansiyeli olan t�m kad�nlarda gebelik testi yap�lmal�d�r.
Karaci�er yetmezli�i:
�iddetli karaci�er yetmezli�i olan hastalarda, orta karaci�er yetmezli�i olan hastalardan elde edilen verilere g�re, niraparib maruziyeti daha y�ksek olabilir ve bu hastalar dikkatlice takip edilmelidir (bkz. B�l�m 4.2 ve 5.2).
Yard�mc� maddeler
Bu t�bbi �r�n laktoz i�ermektedir. Galaktoz intolerans�, total laktaz eksikli�i ya da glukoz- galaktoz malabsorpsiyonu gibi nadir kal�t�msal sorunlar� olan hastalar bu ilac� kullanmamal�d�r.
4.5. Di�er t�bbi �r�nler ile etkile�imler ve di�er etkile�im �ekilleri
Farmakodinamik etkile�imler
Niraparibin a��lar veya imm�nosupresan ajanlarla kombinasyonu incelenmemi�tir.
Niraparibin sitotoksik t�bbi �r�nlerle birlikte kullan�m�yla ili�kili veriler s�n�rl�d�r. Bu nedenle niraparib; a��lar, imm�nosupresan ajanlar veya di�er sitotoksik t�bbi �r�nlerle birlikte kullan�ld��� zaman dikkatli olunmal�d�r.
Farmakokinetik etkile�imler
Di�er t�bbi �r�nlerin niraparibin �zerindeki etkileri CYP substrat� olarak niraparib (CYP1A2 ve CYP3A4)
Niraparib, in vivo karboksilesterazlar (CE) ve UDP-glukuronosil transferazlar�n (UGT) bir
substrat�d�r.
Niraparibin oksidatif metabolizmas� in vivo ortamda minimaldir. CYP enzimlerini inhibisyona u�ratt��� (�rn. itrakonazol, ritonavir ve klaritromisin) veya ind�kledi�i (�rn. rifampin, karbamazepin ve fenitoin) bilinen t�bbi �r�nlerle e�zamanl� olarak uyguland���nda ZEJULA i�in doz ayarlamas� gerekmemektedir.
D��a ak�� ta��y�c�lar�n�n substrat� olarak niraparib (P-gp, BCRP, BSEP, MRP2 ve MATE1/2)
Niraparib, P-glikoproteini (P-gp) ve Meme Kanseri Diren� Proteininin (Breast Cancer Resistance Protein) (BCRP) bir substrat�d�r. Ancak, y�ksek ge�irgenli�i ve biyoyararlan�m� nedeniyle, bu ta��y�c�lar� inhibisyona u�ratan t�bbi �r�nlerle klinik a��dan anlaml� etkile�im riski d���kt�r. Bu nedenle, P-gp'yi (�rn. amiodaron, verapamil) veya BCRP'yi (�rn. osimertinib, velpatasvir ve eltrombopag) inhibisyona u�ratt��� bilinen t�bbi �r�nlerle e�zamanl� olarak uyguland���nda ZEJULA i�in doz ayarlamas� gerekmemektedir.
Niraparib safra tuzu eksport pompas� (substrate of bile salt export pump) (BSEP) veya �oklu ila� direnciyle ili�kili protein 2'nin (MRP2) bir substrat� de�ildir. Maj�r birincil metabolit olan M1, P-gp, BCRP, BSEP veya MRP2'nin bir substrat� de�ildir. Niraparib �oklu ila� ve toksin ekstr�zyonu (MATE)-1 veya 2'nin substrat� de�ilken, M1 bunlar�n ikisinin de substrat�d�r.
Hepatik al�m ta��y�c�lar�n�n substrat� olarak niraparib (OATP1B1, OATP1B3 ve OCT1)
Niraparib veya M1 organik anyon ta��y�c� polipeptit 1B1 (OATP1B1), 1B3 (OATP1B3) veya organik katyon ta��y�c� 1'in (OCT1) substrat� de�ildir. OATP1B1 veya 1B3 (�rn. gemfibrozil, ritonavir) veya OCT1 (�rn. dolutegravir) al�m ta��y�c�lar�n� inhibisyona u�ratt��� bilinen t�bbi �r�nlerle e�zamanl� olarak uyguland���nda ZEJULA i�in doz ayarlamas� gerekmemektedir.
Renal al�m ta��y�c�lar�n�n substrat� olarak niraparib (OAT1, OAT3 ve OCT2)
Niraparib veya M1 organik anyon ta��y�c�s� 1 (OAT1), 3 (OAT3) ve organik katyon ta��y�c�s� 2'nin (OCT2) substrat� de�ildir. OAT1 (�rn. probenesid) veya OAT3 (�rn. probenesid, diklofenak) veya OCT2 al�m ta��y�c�lar�n� (�rn. simetidin, kinidin) inhibisyona u�ratt��� bilinen t�bbi �r�nlerle e�zamanl� olarak uyguland���nda ZEJULA i�in doz ayarlamas� gerekmemektedir.
Niraparibin di�er t�bbi �r�nler �zerindeki etkisi
CYP'lerin (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 ve CYP3A4)
inhibisyonu
Niraparib veya M1, etkin maddeyi metabolize eden CYP enzimlerinin (CYP1A1/2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 ve CYP3A4/5) herhangi birinin inhibit�r� de�ildir.
CYP3A4'�n karaci�erde inhibisyonu beklenmese de, ilgili niraparib konsantrasyonlar�nda CYP3A4'�n ba��rsak d�zeyinde inhibisyon potansiyeli kan�tlanmam��t�r. Bu nedenle, niraparibin metabolizmas�n�n CYP3A4'e ba��ml� oldu�u ve �zellikle dar terap�tik aral��a sahip etkin maddelerle (�rn. siklosporin, takrolimus, alfentanil, ergotamin, pimozid, ketiapin ve halofantrin) kombinasyon halinde kullan�ld��� durumlarda dikkat edilmesi �nerilmektedir.
UDP-glukuronosil transferazlar�n (UGT) inhibisyonu
Niraparib in vitro 200 µM'a kadar dozlarda UGT izoformlar�na kar�� (UGT1A1, UGT1A4, UGT1A9 ve UGT2B7) inhibit�r etki g�stermemi�tir. Bu nedenle, UGT'lerin niraparib taraf�ndan klinik a��dan ilgili inhibisyona u�rat�lma potansiyeli minimaldir.
CYP'lerin ind�ksiyonu (CYP1A2 ve CYP3A4)
Niraparib veya M1 in vitro CYP3A4 ind�kleyicisi de�ildir. �n vitro, niraparib y�ksek konsantrasyonlarda CYP1A2'yi zay�f �ekilde ind�klemektedir ve bu etkinin klinik �nemi tamamen d��lanamamaktad�r. M1 bir CYP1A2 ind�kleyicisi de�ildir. Bu nedenle niraparibin metabolizmas� CYP1A2'ye ba��ml� ve �zellikle terap�tik aral��� dar olan (�rn. klozapin, teofilin ve ropinirol) etkin maddelerle kombinasyon halinde dikkatli kullan�lmas� �nerilmektedir.
D��a ak�� ta��y�c�lar�n�n inhibisyonu (P-gp, BCRP, BSEP, MRP2 ve MATE1/2)
Niraparib, BSEP veya MRP2'nin inhibit�r� de�ildir. �n vitro, niraparib P-gp'yi �ok zay�f d�zeyde ve BCRP'yi s�ras�yla IC= 161 µM ve 5,8 µM de�erlerinde inhibe eder. Bu nedenle, bu d��a ak�� ta��y�c�lar�n�n inhibisyona u�ramas�yla ili�kili klinik a��dan anlaml� bir etkile�im olas�l��� d���k olsa da g�z ard� edilemez. Niraparibin BCRP substratlar�yla (irinotekan, rosuvastatin, simvastatin, atorvastatin, metotreksat) kombinasyon halinde kullan�ld��� durumlarda dikkat edilmesi �nerilmektedir.
Niraparib, MATE1 ve 2'nin inhibit�r�d�r ve ICde�erleri s�ras�yla 0,18 µM ve ≤0,14 µM'dir. Bu ta��y�c�lar�n substratlar� olan t�bbi �r�nlerle (�rn. metformin) birlikte uyguland���nda bu �r�nlerin plazma konsantrasyonlar�nda art�� olas�l��� g�z ard� edilemez.
Maj�r primer metabolit M1, P-gp, BCRP, BSEP, MRP2 veya MATE1/2'nin inhibit�r� gibi
g�r�lmemektedir.
Hepatik al�m ta��y�c�lar�n�n inhibisyonu (OATP1B1, OATP1B3 ve OCT1)
Niraparib veya M1 organik anyon ta��y�c�s� polipeptit 1B1 (OATP1B1) veya 1B3'�n (OATP1B3) inhibit�r� de�ildir.
�n vitro, niraparib organik katyon ta��y�c�s� 1'i (OCT1) IC= 34,4 µM de�eriyle zay�f d�zeyde inhibisyona u�ratmaktad�r. Niraparibin metformin gibi OCT1 yoluyla al�m ta��n�m�na maruz kalan etkin maddelerle kombinasyon halinde kullan�ld��� durumlarda dikkat edilmesi �nerilmektedir.
Renal al�m ta��y�c�lar�n�n inhibisyonu (OAT1, OAT3 ve OCT2)
Niraparib veya M1, organik anyon ta��y�c�s� 1 (OAT1), 3 (OAT3) ve organik katyon ta��y�c�s� 2'yi (OCT2) inhibisyona u�ratmaz.
T�m klinik �al��malar yaln�zca yeti�kinler �zerinde y�r�t�lm��t�r.
�zel pop�lasyona ili�kin ek bilgiler
Veri bulunmamaktad�r.
Pediyatrik pop�lasyon
T�m klinik �al��malar yaln�zca yeti�kinlerde ger�ekle�tirilmi�tir.
4.6. Gebelik ve laktasyon
Gebelik kategorisi: X
�ocuk do�urma potansiyeli bulunan kad�nlar/Do�um kontrol� (Kontrasepsiyon)
�ocuk do�urma potansiyeli olan kad�nlar tedavi s�ras�nda gebe kalmamal� ve tedavi ba�lang�c�nda gebe olmamal�d�r. Tedaviden �nce �ocuk do�urma potansiyeli olan t�m kad�nlarda gebelik testi yap�lmal�d�r. �ocuk do�urma potansiyeli olan kad�nlar tedavi s�ras�nda ve son ZEJULA dozundan sonra 6 ay boyunca y�ksek etkili do�um kontrol y�ntemleri kullanmal�d�r.
Gebelik d�nemi
Niraparibin gebe kad�nlarda kullan�m�yla ilgili veri bulunmamaktad�r veya s�n�rl� miktardad�r. Hayvanlarda �reme veya geli�im toksisitesi �al��mas� yap�lmam��t�r. Ancak, etki mekanizmas�na g�re, niraparib gebe kad�nlara uyguland���nda embriyo-letal ve teratojenik etkiler dahil olmak �zere embriyonik veya fetal hasara yol a�abilir. ZEJULA gebelik s�ras�nda kullan�lmamal�d�r.
Laktasyon d�nemi
Niraparib veya metabolitlerinin insan s�t�ne ge�ip ge�medi�i bilinmemektedir. ZEJULA uygulamas� s�ras�nda ve son dozun al�nmas�ndan sonraki 1 ay boyunca emzirme kontrendikedir (bkz. B�l�m 4.3).
�reme yetene�i/Fertilite
Fertiliteyle ilgili klinik veri bulunmamaktad�r. S��anlarda ve k�peklerde spermatojenezde reversibl bir d���� g�zlemlenmi�tir (bkz. B�l�m 5.3).
4.7. Ara� ve makine kullan�m� �zerindeki etkiler
ZEJULA'nun ara� veya makine kullan�m� �zerindeki etkisi orta d�zeydedir. ZEJULA alan hastalarda asteni, halsizlik, ba� d�nmesi ya�ayabilir veya konsantrasyon g��l��� g�r�lebilir. Bu semptomlar� ya�ayan hastalar ara� veya makine kullan�rken dikkat etmelidir.
4.8. �stenmeyen etkiler
G�venlilik profilinin �zeti
Havuzlanm�� PRIMA (ba�lang�� dozu 200 mg veya 300 mg) ve NOVA �al��malar�nda ZEJULA monoterapisi alan 851 hastan�n ≥%10'unda g�r�len t�m derecelerdeki advers reaksiyonlar (ADR) bulant�, anemi, trombositopeni, halsizlik, kab�zl�k, kusma, ba� a�r�s�, insomnia, trombosit say�m�nda d����, n�tropeni, kar�n a�r�s�, i�tah azalmas�, diyare, dispne, hipertansiyon, asteni, ba� d�nmesi, n�trofil say�m�nda d����, �ks�r�k, artralji, s�rt a�r�s�, beyaz kan h�cresi say�m�nda d���� ve s�cak basmas�d�r.
En yayg�n ciddi advers reaksiyonlar %1'den fazla (tedavide ortaya ��kan s�kl�klar) trombositopeni ve anemidir.
Advers reaksiyonlar�n tablolanm�� listesi
ZEJULA monoterapisi alan hastalarda klinik ara�t�rmalara ve pazarlama sonras� g�zleme dayal� olarak a�a��daki advers reaksiyonlar g�r�lm��t�r (bkz. Tablo 4). Advers reaksiyonlar�n g�r�lme s�kl�klar�, hasta maruziyetinin bilindi�i PRIMA ve NOVA klinik �al��malar�ndan (300 mg/g�n'l�k sabit ba�lang�� dozu) elde edilen havuzlanm�� advers olay verilerine dayanmaktad�r ve �u �ekilde tan�mlanm��t�r: �ok yayg�n (≥1/10); yayg�n (≥1/100 ila <1/10); yayg�n olmayan (≥1/1.000 ila <1/100); seyrek (≥1/10.000 ila <1/1.000); �ok seyrek (<1/10.000); bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor). Her bir s�kl�k gruplamas�nda, istenmeyen etkiler, azalan ciddiyet s�ras�na g�re bildirilmektedir.
Tablo 4: Advers reaksiyonlar�n tablolanm�� listesi
Sistem Organ S�n�f� | T�m CTCAE* derecelerinin s�kl��� | 3. veya 4. CTCAE* derecelerinin s�kl��� |
Enfeksiyonlar ve enfestasyonlar | �ok yayg�n �drar yolu enfeksiyonu Yayg�n Bron�it, konjonktivit | Yayg�n olmayan �drar yolu enfeksiyonu, bron�it |
(Kist ve polipler de dahil olmak �zere) iyi huylu, k�t� huylu ve tan�mlanmam�� neoplazmalar | Yayg�n Miyelodisplastik sendrom/ akut miyeloid l�semi** | Yayg�n Miyelodisplastik sendrom/ akut miyeloid l�semi** |
Kan ve lenf sistemi hastal�klar� | �ok yayg�n Trombositopeni, anemi, n�tropeni, l�kopeni Yayg�n olmayan Pansitopeni, febril n�tropeni | �ok yayg�n Trombositopeni, anemi, n�tropeni Yayg�n L�kopeni Yayg�n olmayan Pansitopeni, febril n�tropeni |
Ba����kl�k sistemi hastal�klar� | Yayg�n A��r� duyarl�l�k | Yayg�n olmayan A��r� duyarl�l�k |
Metabolizma ve beslenme hastal�klar� | �ok yayg�n ��tah azalmas� Yayg�n Hipokalemi | Yayg�n Hipokalemi Yayg�n olmayan ��tah azalmas� |
Psikiyatrik hastal�klar | �ok yayg�n Insomnia Yayg�n Anksiyete, depresyon, bili�sel bozukluk†† Yayg�n olmayan Konf�zyon | Yayg�n olmayan Insomnia, anksiyete, depresyon, konf�zyon |
Sistem Organ S�n�f� | T�m CTCAE* derecelerinin s�kl��� | 3. veya 4. CTCAE* derecelerinin s�kl��� |
Sinir sistemi hastal�klar� | �ok yayg�n Ba� a�r�s�, ba� d�nmesi Yayg�n Tat alma duygusunda bozukluk (disg�zi) Seyrek Posterior Reversibl Ensefalopati Sendromu (PRES)** | Yayg�n olmayan Ba� a�r�s� |
Kardiyak hastal�klar | �ok yayg�n Palpitasyon Yayg�n Ta�ikardi |
|
Vask�ler hastal�klar | �ok yayg�n Hipertansiyon Seyrek Hipertansif kriz | Yayg�n Hipertansiyon |
Solunum, g���s bozukluklar� ve mediastinal hastal�klar | �ok yayg�n Dispne, �ks�r�k, nazofarenjit Yayg�n Epistaksis Yayg�n olmayan Pn�monit | Yayg�n olmayan Dispne, epistaksis, pn�monit |
Gastrointestinal hastal�klar | �ok yayg�n Bulant�, kab�zl�k, kusma, kar�n a�r�s�, diyare, dispepsi Yayg�n A��z kurulu�u, abdominal distensiyon, mukozal enflamasyon (mukoza iltihab�), stomatit | Yayg�n Bulant�, kusma, kar�n a�r�s� Yayg�n olmayan Diyare, kab�zl�k, mukozal enflamasyon (mukoza iltihab�), stomatit, a��z kurulu�u |
Deri ve deri alt� doku hastal�klar� | Yayg�n Fotoduyarl�l�k, d�k�nt� | Yayg�n olmayan Fotoduyarl�l�k, d�k�nt� |
Kas-iskelet bozukluklar�, ba� doku ve kemik hastal�klar� | �ok yayg�n S�rt a�r�s�, artralji Yayg�n Miyalji | Yayg�n olmayan S�rt a�r�s�, artralji, miyalji |
Genel bozukluklar ve uygulama b�lgesine ili�kin hastal�klar� | �ok yayg�n Halsizlik, asteni Yayg�n Periferik �dem | Yayg�n Halsizlik, asteni |
Sistem Organ S�n�f� | T�m CTCAE* derecelerinin s�kl��� | 3. veya 4. CTCAE* derecelerinin s�kl��� |
Ara�t�rmalar | Yayg�n Gamma-glutamil transferaz art���, AST art���, kan kreatinin art���, ALT art���, kan alkalin fosfataz art���, v�cut a��rl���nda azalma | Yayg�n Gamma-glutamil transferaz art���, ALT art��� Yayg�n olmayan AST art���, kan alkalin fosfataz art��� |
* CTCAE = Advers Olaylar ��in Ortak Terminoloji Kriterleri versiyon 4.02.
** Niraparib klinik �al��ma verilerine dayanmaktad�r. Pivot ENGOT-OV16 monoterapi �al��mas�yla s�n�rl� de�ildir.
�rtiker dahildir.
††Haf�za bozuklu�u, konsantrasyon bozuklu�u i�erir.
Bazal a��rl�k veya trombosit say�m�na g�re 200 mg ZEJULA ba�lang�� dozu uygulanan hasta grubunda g�r�len advers reaksiyonlar, sabit ba�lang�� dozu 300 mg uygulanan grupla kar��la�t�r�ld���nda benzer veya daha d���k s�kl�kta g�r�lm��t�r (Tablo 4).
Trombositopeni, anemi ve n�tropeni s�kl���yla ilgili �zg�n bilgiler i�in a�a��daki b�l�me bak�n�z.
Se�ili advers reaksiyonlar�n tan�m�
Klinik tan�lar ve/veya laboratuvar bulgular� dahil olmak �zere hematolojik advers reaksiyonlar (trombositopeni, anemi, n�tropeni) genellikle niraparib tedavisinin erken d�nemlerinde ger�ekle�mi� ve zaman i�inde g�r�lme oranlar� azalm��t�r.
NOVA ve PRIMA �al��malar�nda, ZEJULA tedavisine uygun olan hastalar�n bazal hematolojik parametreleri ��yledir: Tedaviden �nce mutlak n�trofil say�s� (MNS) ≥1.500 h�cre/µL; trombosit ≥100,000 h�cre/µL ve hemoglobin ≥9 g/dL (NOVA) veya ≥10 g/dL (PRIMA). Klinik programda, hematolojik advers reaksiyonlar laboratuvar izlem ve doz de�i�iklikleriyle y�netilmi�tir (bkz. B�l�m 4.2).
PRIMA'da, bazal a��rl�k veya trombosit say�m�na g�re ZEJULA ba�lang�� dozu uygulanan hastalarda, ≥3. derece trombositopeni, anemi ve n�tropeni, 300 mg sabit ba�lang�� dozu verilen grupla kar��la�t�r�ld���nda s�ras�yla %48'den %21'e, %36'dan %23'e, %24'ten %15'e d��m��t�r. Hastalar�n s�ras�yla %3, %3 ve %2'sinde trombositopeni, anemi ve n�tropeni nedeniyle tedavi b�rak�lm��t�r.
Trombositopeni
PRIMA'da, ZEJULA ile tedavi edilen hastalar�n %39'unda, plaseboyla tedavi edilen hastalar�n
%0,4'�nde 3./4. derece trombositopeni g�r�lm��t�r ve ilk dozdan ilk ba�lang�ca kadar ge�en medyan s�re 22 g�n (aral�k: 15 ila 335 g�n), medyan s�re 6 g�nd�r (aral�k: 1 ila 374 g�n). Niraparib alan hastalar�n %4'�nde trombositopeni nedeniyle tedavi b�rak�lm��t�r.
NOVA'da, ZEJULA alan hastalar�n yakla��k %60'�nda herhangi bir derecede trombositopeni ve hastalar�n %34'�nde 3./4. derece trombositopeni g�r�lm��t�r. Bazal trombosit say�m� 180 × 10/L'den d���k olan hastalarda, herhangi bir derecede ve 3./4. derece trombositopeni hastalar�n s�ras�yla %76's� ve %45'inde ger�ekle�mi�tir. Derecesinden ba��ms�z olarak trombositopeni ve
/4. derece trombositopeninin ba�lang�c�na kadar ge�en medyan s�re s�ras�yla 22 ve 23 g�nd�r.
d�ng�den itibaren tedavinin ilk 2 ay�nda yap�lan yo�un doz de�i�ikliklerinden sonra yeni trombositopeni g�r�lme oran� %1,2'dir. Herhangi bir derecedeki trombositopeni olaylar�n�n medyan s�resi 23 g�nd�r ve 3./4. derece trombositopeni olaylar�n�n medyan s�resi 10 g�nd�r. ZEJULA ile tedavi edilen hastalarda hemoraji riski y�kselebilir. Klinik programda, trombositopeni laboratuvar izlemi, doz modifikasyonu ve uygun olan yerlerde trombosit transf�zyonuyla y�netilmi�tir (bkz. B�l�m 4.2). Hastalar�n yakla��k %3'� trombositopeni olaylar� (trombositopeni ve trombosit say�m�nda d����) nedeniyle tedaviyi b�rakm��t�r.
4.9. Doz a��m� ve tedavisi
ZEJULA doz a��m� durumu i�in spesifik bir tedavi yoktur ve doz a��m� semptomlar� belirlenmemi�tir. Doz a��m� durumunda, hekimler genel destekleyici �nlemleri izlemeli ve semptomatik tedavi uygulamal�d�r.
5. FARMAKOLOJ�K �ZELL�KLER
5.1. Farmakodinamik �zellikler
Farmakoterap�tik grup: Di�er antineoplastik ajanlar, Poli (ADP-riboz) polimeraz (PARP) inhibit�rleri
ATC kodu: L01XK02.
Etki mekanizmas� ve farmakodinamik etkiler
Niraparib, DNA onar�m�nda rol oynayan poli(ADP-riboz) polimeraz (PARP) enzimleri PARP- 1 ve PARP-2'nin bir inhibit�r�d�r. �n vitro �al��malarda, niraparible ind�klenen sitotoksisiteye PARP enzimatik aktivitesinin inhibisyonu ve DNA hasar�, apoptoz ve h�cre �l�m� ile sonu�lanan PARP-DNA komplekslerinin olu�umunda art���n dahil olabilece�i g�r�lm��t�r. Meme kanseri (BRCA) 1 ve 2 t�m�r bask�lay�c� genlerinde eksiklik olan veya olmayan t�m�r h�cre hatlar�nda artan niraparib kaynakl� sitotoksisite g�zlenmi�tir.Farelerde b�y�t�len
ortotopik y�ksek dereceli ser�z over kanseri hasta kaynakl� ksenograft t�m�rlerinde (PDX), niraparibin BRCA 1 ve 2 mutantlar�, homolog rekombinasyon (HR) eksikli�i olan BRCA vah�i tip ve tespit edilebilir HR eksikli�i olmayan BRCA vah�i tip t�m�rlerde t�m�r b�y�mesini azaltt��� g�r�lm��t�r.
Klinik etkililik ve g�venlilik
Birinci basamak over kanseri idame tedavisi
PRIMA, Faz 3 �ift k�r, plasebo kontroll� bir �al��mad�r ve birinci basamak platin bazl� kemoterapiye tam veya k�smi yan�t veren hastalar (n=733) 2:1 oran�nda ZEJULA veya e�le�tirilmi� plaseboya randomize edilmi�tir. PRIMA, 475 hastada (317'si niraparib koluna, 158'i plasebo koluna randomize edilmi�tir) devam eden 28 g�nl�k d�ng�lerde g�nde tek doz 300 mg ba�lang�� dozuyla ba�lat�lm��t�r. PRIMA'n�n ba�lang�� dozu Protokol�n 2. De�i�ikli�i ile de�i�tirilmi�tir. Bu noktadan sonra, bazal v�cut a��rl��� ≥77 kg ve bazal trombosit say�m� ≥ 150.000/µL olan hastalara g�nl�k ZEJULA 300 mg (n=34) veya plasebo (n=21) tedavisi uygulan�rken, bazal v�cut a��rl��� <77 kg ve bazal trombosit say�m� < 150.000/µL olan hastalara g�nl�k ZEJULA 200 mg (n=122) veya plasebo (n=61) tedavisi uygulanm��t�r.
Hastalar birinci basamak platin bazl� kemoterapi yan� s�ra ameliyat oldu ise, ameliyat�n tamamlanmas�ndan sonra randomize edilmi�tir. G�n�ll�ler kemoterapinin son d�ng�s�n�n ilk g�n�nden sonraki 12 hafta i�inde randomize edilmi�tir. G�n�ll�lere ≥6 ve ≤9 platin bazl� tedavi d�ng�s� uygulanm��t�r. T�m�r hacminin azalt�ld��� ara ameliyatlardan sonra, g�n�ll�lere ≥2 postoperatif platin bazl� tedavi d�ng�s� uygulanm��t�r. Kemoterapiyle birlikte bevasizumab verilen ama idame tedavisi olarak bevasizumab alamayan hastalar �al��ma d���nda tutulmam��t�r. Hastalar ZEJULA dahil olmak �zere ge�mi� PARP inhibit�r� (PARPi) tedavisi g�rmemi� olmal�d�r. Neoadjuvan kemoterapi ve sonras�nda t�m�r hacminin azalt�ld��� ara ameliyat ge�iren hastalarda g�r�n�r rezid�el hastal�k olabilir veya rezid�el hastal�k olmayabilir. Hastal��� 3. evrede olan ve t�m�r hacminin azalt�ld��� primer ameliyattan sonra tam sitored�ksiyon uygulanan (yani g�r�n�r rezid�el hastal��� olmayan) hastalar �al��man�n d���nda b�rak�lm��t�r. Randomizasyon, birinci basamak platin rejimi s�ras�nda verdi�i en iyi yan�t (tam yan�t ya da k�smi yan�t), neoadjuvan kemoterapi (NAKT) (Evet ya da Hay�r) ve homolog rekombinasyon eksikli�i (HRD) durumuna [pozitif (HR eksikli�i) ya da negatif (HR eksikli�i yok) veya belirlenememi�] g�re tabakala�t�r�lm��t�r. HRD testi, ilk tan� tarihinde al�nan t�m�r dokusunda yap�lan HRD testi kullan�larak yap�lm��t�r. CA-125 d�zeyleri hastan�n birinci basamak tedavisi s�ras�nda normal aral�kta (veya >%90 CA-125 d�����) olmal� ve en az 7 g�n boyunca stabil devam etmelidir.
Hastalar 1. d�ng�/1. g�nde (C1/D1), devaml� 28 g�nl�k d�ng�lerde g�nde tek doz uygulanan ZEJULA 200 veya 300 mg veya kar��l�k gelen plasebo ile tedaviyle ba�lam��t�r. Her bir d�ng�de klinik ziyaretleri ger�ekle�tirilmi�tir (4 hafta ±3 g�n).
Birincil sonlan�m noktas�, RECIST'e (versiyon 1.1) k�r ba��ms�z merkezi inceleme (BICR) ile belirlenen progresyonsuz (ilerlemesiz) sa�kal�md�r (PFS). Genel sa�kal�m (OS) anahtar ikincil hedeftir. PFS testi, ilki HR eksikli�i olan pop�lasyonda ve sonra genel pop�lasyonda olmak �zere hiyerar�ik olarak yap�lm��t�r: Medyan ya� 62'dir ve ZEJULA'ya randomize edilen hastalarda 32 ila 85 ya� aral���nda, plaseboyla randomize edilen hastalarda 33 ila 88 ya� aral���ndad�r. T�m hastalar�n %89'u beyazd�r. ZEJULA ile randomize edilen hastalar�n %69'u, plaseboyla tedavi edilen hastalar�n %71'inin �al��man�n ba�lang�c�ndaki ECOG skoru 0'd�r. Genel pop�lasyonda, hastalar�n %65'inde 3. evre hastal�k, %35'inde 4. evre hastal�k vard�r. Genel pop�lasyonda, �o�u hastan�n (≥%80) primer t�m�r b�lgesi overdir; �o�u hastada (>%90) ser�z histolojiye sahip t�m�r vard�r. Hastalar�n %67'si neoadjuvan kemoterapi NAKT alm��t�r. Hastalar�n %69'u birinci basamak platin bazl� kemoterapiye tam yan�t vermi�tir. Niraparib verilen toplam 6 hasta over kanseri i�in �nceden bevasizumab tedavisi g�rm��t�r.
PRIMA �al��mas�nda, HR (Homolog Rekombinasyonu) eksikli�i (HRD) olan ve genel pop�lasyonda plaseboyla kar��la�t�r�ld���nda ZEJULA'ya randomize edilen hastalarda PFS'de istatistiksel a��dan anlaml� iyile�me g�r�lm��t�r (Tablo 5, �ekil 1 ve 2).
�kincil etkililik sonlan�m noktalar� aras�nda, ilk izleyen tedaviden sonra PFS (PFS2) ve OS bulunmaktad�r (Tablo 5).
Tablo 5: Etkililik sonu�lar� - PRIMA (BICR belirlemesine g�re)
| HR eksikli�i olan pop�lasyon | Genel pop�lasyon | ||
ZEJULA (N=247) | plasebo (N=126) | ZEJULA (N=487) | plasebo (N=246) | |
Medyan PFS (%95 GA) | 21,9 (19,3; NE) | 10,4 (8,1; 12,1) | 13,8 (11,5; 14,9) | 8,2 (7,3; 8,5) |
Tehlike oran� (%95 GA) | 0,43 (0,31; 0,59) | 0,62 (0,5; 0,76) | ||
p de�eri | <0,0001 | <0,0001 | ||
PFS2 Tehlike oran� (%95 GA) | 0,84 (0,485; 1,453) | 0,81 (0,577; 1,139) | ||
OS* Tehlike oran� (%95 GA) | 0,61 (0,265; 1,388) | 0,7 (0,44; 1,11) | ||
PFS = progresyonsuz sa�kal�m; GA = g�ven aral���; NE = de�erlendirilemez; OS = genel sa�kal�m; PFS2 = sonraki ilk tedaviden sonra progresyonsuz sa�kal�m.
* Primer PFS analizi tarihinde, genel pop�lasyonda ZEJULA alan hastalar�n %84'�nde, plasebo alan hastalar�n %77'sinde randomizasyondan iki y�l sonra tahmini sa�kal�m.
PFS2 ve OS verileri hen�z olgunla�mam��t�r.
�ekil 1: HR eksikli�i olan t�m�rlere sahip hastalarda progresyonsuz sa�kal�m -
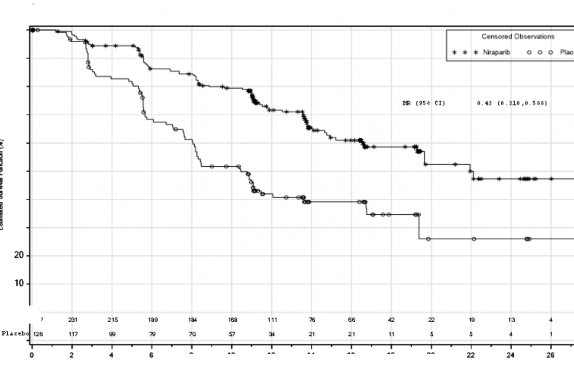
PRIMA(ITT pop�lasyonu, N=373)
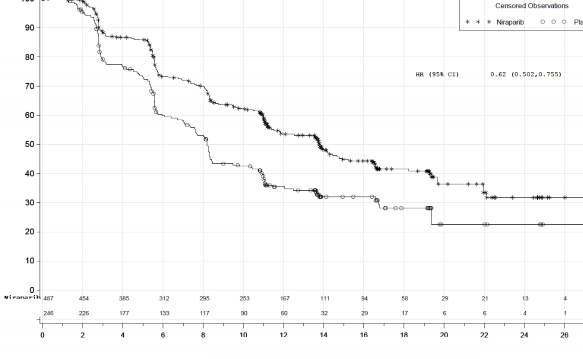
�ekil 2: Genel pop�lasyonda progresyonsuz sa�kal�m - PRIMA (ITT pop�lasyonu, N=733)
Alt grup analizleri
Homolog Rekombinasyon (HR) eksikli�i olan pop�lasyonda, BRCA (+) over kanserli hastalardan olu�an alt grupta 0,4 tehlike oran� (%95 GA 0,27; 0,62) g�zlenmi�tir (N=223). HRD BRCA (-) hastalardan olu�an alt grupta (N=150), 0,5 tehlike oran� (%95 GA 0,31; 0,83) g�zlemlenmi�tir. Homolog Rekombinasyon eksikli�i olmayan pop�lasyonda (N=249); 0,68 tehlike oran� (%95 GA 0,49; 0,94) g�zlenmi�tir.
Bazal v�cut a��rl��� veya trombosit say�m�na g�re 200 veya 300 mg ZEJULA uygulanan hastalar�n ke�ifsel alt grup analizlerinde, benzer etkililik g�zlenmi�tir (ara�t�rmac�n�n de�erlendirdi�i PFS) ve tehlike oran� HRD olan pop�lasyonda 0,54 (%95 GA 0,33; 0,91) ve genel pop�lasyonda 0,68'dir (%95 GA 0,49; 0,94). HRD olmayan alt grupta, 200 mg dozunun tedavi etkisinin 300 mg dozuyla kar��la�t�r�ld���nda daha d���k oldu�u g�r�lm��t�r.
Platine duyarl� rek�rren (tekrarlayan) over kanseri idame tedavisi
�dame tedavisi olarak niraparib g�venlili�i ve etkilili�i, n�kseden a��rl�kl� olarak y�ksek derecede ser�z epitelyal over, fallop t�p� veya birincil periton kanseri hastas� olan ve alt� aydan uzun s�re sondan �nceki platin bazl� tedavilerine kadar tam yan�t (CR) veya k�smi yan�t (PR) tan�mlar�na g�re platin duyarl� hastalarda ger�ekle�tirilen Faz 3 randomize, �ift k�r, plasebo kontroll� uluslararas� bir �al��mada (NOVA) incelenmi�tir. Hastalar�n niraparib tedavisine uygun olmas� i�in son platin temelli kemoterapinin tamamlanmas�ndan sonra yan�t vermesi (CR veya PR) gerekmektedir. Son platin tedavilerinin akabinde CA-125 seviyeleri normal (veya ba�lang��tan CA-125'te >%90'l�k bir d����) olmal�d�r ve en az 7 g�n s�re ile stabil kalmal�d�r. Hastalar daha �nce ZEJULA dahil olmak �zere hi�bir PARPi tedavisi almam�� olmal�d�r. Uygun hastalar germline BRCA (gBRCA) mutasyon testinin sonucuna g�re iki kohorttan birine atanm��t�r. Her kohort i�inde, hastalar 2:1 oran�nda niraparib ve plaseboya randomize edilmi�tir. Hastalar randomizasyondan �nce gBRCA analizi i�in al�nan kan numunelerine g�re gBRCA mutasyonlu kohortuna atanm��t�r. T�m�r BRCA (tBRCA) mutasyonu ve HRD testleri,
ilk tan� kondu�u tarihte veya tekrar ger�ekle�me tarihinde al�nan t�m�r dokusu �zerinde yap�lan HRD testi kullan�larak ger�ekle�tirilmi�tir.
T�m kohortlardaki randomizasyon, �al��maya kat�lmadan �nceki platin tedavisinden sonra ilerlemeye kadar ge�en zamana (6 ila <12 ay ve ≥12 ay); sondan �nceki veya son platin rejimiyle birlikte bevasizumab kullan�lmad���na ve en yak�n ge�mi�teki platin rejimi s�ras�ndaki en iyi yan�ta (tam yan�t ve k�smi yan�t) g�re tabakala�t�r�lm��t�r.
D�ng� 1/1.g�ndeki (C1/D1) hastalar, 28 g�nl�k s�rekli d�ng�ler halinde g�nde tek doz uygulanan niraparib 300 mg veya e�le�tirilmi� plasebo ile tedaviye ba�lam��t�r. Her bir d�ng�de klinik ziyaretleri yap�lm��t�r (4 hafta ± 3 g�n).
NOVA �al��mas�nda, 1. d�ng�de hastalar�n %48'inin dozuna ara verilmi�tir. Hastalar�n yakla��k %47'si 2. d�ng�de daha d���k bir dozla tekrar ba�lam��t�r.
NOVA �al��mas�nda niraparible tedavi edilen hastalarda en yayg�n olarak kullan�lan doz 200 mg'd�r.
Progresyonsuz sa�kal�m (PFS), RECIST 1'e (Solid T�m�rlerde Yan�t De�erlendirme, versiyon 1.1) veya klinik belirtiler ve semptomlara ve CA-125 art���na g�re belirlenmi�tir. PFS, (kemoterapi rejiminin tamamlanmas�ndan 8 haftaya kadar sonraki) randomizasyon tarihinden hastal�k ilerlemesi veya �l�me kadar �l��lm��t�r.
PFS i�in primer etkililik analizi, k�rle�tirilmi� merkezi ba��ms�z de�erlendirmeyle belirlenmi�tir ve gBRCA mutasyonlu kohort ve gBRCA mutasyonsuz kohort i�in ayr�ca ileriye d�n�k �ekilde tan�mlanm�� ve de�erlendirilmi�tir. Genel sa�kal�m (OS) analizleri, ikincil sonu� �l��tleridir.
�kincil etkililik sonlan�m noktalar� aras�nda, kemoterapisiz ara (CFI), izleyen ilk tedaviye kadar ge�en s�re (TFST), izleyen ilk tedaviden sonra ge�en s�re (PFS2) ve OS'dir (genel sa�kal�m).
gBRCA mutasyonlu (n=203) ve gBRCA mutasyonsuz kohortlarda (n=350) niraparib ve plasebo kollar� aras�nda demografik, bazal hastal�k karakteristikleri ve ge�mi� tedavi �yk�s� genellikle dengelidir. Medyan ya�lar t�m tedaviler ve kohortlarda 57 ila 63 aral���ndad�r. T�m kohortlarda �o�u hastadaki (>%80) birincil t�m�r b�lgesi overlerdir; �o�u hastada (>%84) ser�z histolojiye sahip t�m�rler vard�r. Her iki kohorttaki her iki tedavi kolunda bulunan hastalar�n y�ksek bir oran� daha �nce 3 veya daha fazla kemoterapi basama�� alm��t�r; bunlara gBRCA mutasyonlu ve gBRCA mutasyonsuz kohortlardaki niraparib hastalar�n�n %49 ve %34'� dahildir. �o�u hasta 18 ila 64 ya��ndad�r (%78), beyazd�r (%86) ve ECOG performans skoru 0'd�r (%68).
gBRCA mutasyonlu kohortta, tedavi d�ng�s� medyan say�s� niraparib kolunda plasebo koluna g�re daha y�ksektir (s�ras�yla 14 ve 7 d�ng�). Niraparib grubunda plasebo grubuyla kar��la�t�r�ld���nda daha fazla say�da hasta (s�ras�yla %54,4 ve %16,9) 12 aydan uzun s�re tedaviye devam etmi�tir.
Genel gBRCA mutasyonsuz kohortta, tedavi d�ng�lerinin medyan say�s� niraparib kolunda plasebo koluna g�re daha y�ksektir (s�ras�yla 8 ve 5 d�ng�). Niraparib grubunda plasebo grubuyla kar��la�t�r�ld���nda daha fazla say�da hasta (s�ras�yla %34,2 ve %21,1) 12 aydan uzun s�re tedaviye devam etmi�tir.
�al��ma gBRCA mutasyonlu kohortta ve gBRCA mutasyonsuz kohortta plaseboyla kar��la�t�r�ld���nda niraparib idame tedavisi i�in PFS'de istatistiksel a��dan anlaml� iyile�me olan esas amac�na ula�m��t�r. Tablo 6 ve �ekil 3'te, primer etkililik pop�lasyonlar�nda PFS birincil sonlan�m noktas� i�in sonu�lar g�r�lmektedir (gBRCA mutasyonlu kohort ve genel gBRCA mutasyonsuz kohort).
Tablo 6: NOVA �al��mas�ndaki birincil nesnel sonu�lar�n �zeti
| gBRCA mutasyonlu kohort | gBRCA mutasyonsuz kohort | ||
| Niraparib (N=138) | plasebo (N=65) | niraparib (N=234) | plasebo (N=116) |
PFS medyan (%95 GA*) | 21 (12,9; NR) | 5,5 (3,8; 7,2) | 9,3 (7,2; 11,2) | 3,9 (3,7; 5,5) |
P de�eri | <0,0001 | <0,0001 | ||
Tehlike oran� (TO) (Niraparib:plasebo) (%95 GA*) | 0,27 (0,173; 0,41) | 0,45 (0,338; 0,607) | ||
PFS = progresyonsuz sa�kal�m; GA = g�ven aral���; NE = de�erlendirilemez
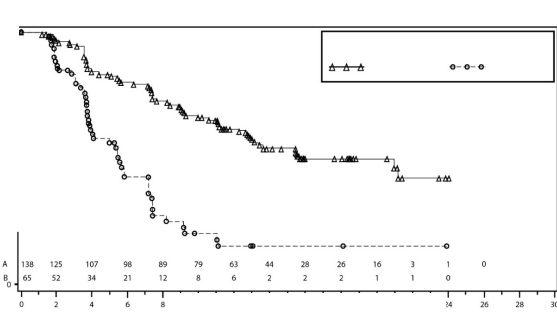
�ekil 3: IRC de�erlendirmesine g�re gBRCA mutasyonlu kohortta progresyonsuz sa�kal�m i�in Kaplan-Meier grafi�i - NOVA (ITT pop�lasyonu, N=203)
Estimated Survival Function
Time since Randomization (Months)
�ekil 4: IRC de�erlendirmesine g�re genel olarak gBRCA mutasyonsuz kohortta progresyonsuz sa�kal�m i�in Kaplan-Meier grafi�i - NOVA (ITT pop�lasyonu, n=350)
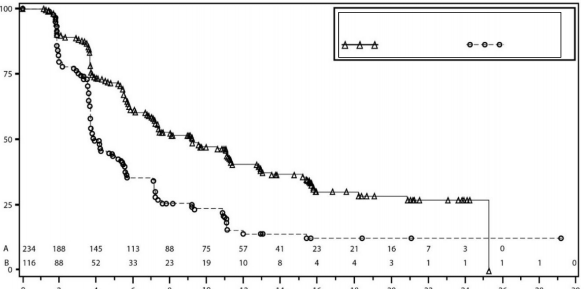
NOVA'da ikincil etkililik sonlan�m noktalar�
Son analizde, gBRCAmut kohortunda medyan PFS2, niraparib ile tedavi edilen hastalarda 29,9 ay iken, plasebo alan hastalarda 22,7 ayd�r (HR = 0,70; % 95 GA: 0,50, 0,97). gBRCAmut olmayan kohorttaki medyan PFS2, niraparib ile tedavi edilen hastalarda 19,5 ay iken, plasebo alan hastalarda 16,1 ayd�r (HR = 0,80; %95 GA: 0,63, 1,02).
Genel sa�kal�m�n son analizinde, gBRCAmut kohortunda (n = 203) medyan OS, niraparib ile tedavi edilen hastalarda 40,9 ay, plasebo alan hastalarda ise 38,1 ay (HR = 0,85; %95 GA: 0,61, 1,20) oldu�u g�r�lm��t�r. gBRCAmut kohortu i�in kohort olgunlu�u %76'd�r. gBRCAmut olmayan kohortta (n = 350) medyan OS, niraparib ile tedavi edilen hastalarda 31.0 ay, plasebo alan hastalarda ise 34.8 ay (HR = 1.06; %95 GA: 0.81, 1.37) oldu�u g�r�lm��t�r. gBRCAmut olmayan kohort i�in kohort olgunlu�u %79'dur.
Do�rulanm�� anket y�ntemleriyle (FOSI ve EQ-5D) elde edilen hasta beyanl� sonu� (PRO) verileri, niraparible tedavi edilen hastalar�n ya�am kalitesiyle ili�kilendirilen �l��mlerde plasebodan farkl� olmad���n� g�stermi�tir.
Pediyatrik pop�lasyon
Avrupa �la� Ajans� (EMA), ZEJULA ile over karsinomda (rabdomiyosarkoma ve germ h�creli t�m�rler hari�) pediyatrik pop�lasyon t�m alt gruplar�nda �al��ma sunma y�k�ml�l��� konusunda muafiyet getirmi�tir.
5.2. Farmakokinetik �zellikler
Genel �zelliklerEmilim:
A�l�k ko�ullar�nda tek doz 300 mg niraparib uyguland�ktan sonra, niraparib plazmada 30 dakika i�inde �l��lebilir d�zeydedir ve niraparib i�in ortalama pik plazma konsantrasyonuna (C)
yakla��k 3 saatte ula��lm��t�r [804 ng/mL (%VK: %50,2]. G�nde bir kez 30 mg ila 400 mg aral���nda �oklu oral niraparib dozlar�ndan sonra, niraparib birikimi yakla��k 2 ila 3 katt�r.
Niraparib dozu 30 mg'dan 400 mg'a artt�r�ld���nda niraparibe sistemik maruziyetler (Cve EAA) dozla orant�l� �ekilde artm��t�r. Niraparibin mutlak biyoyararlan�m� yakla��k %73't�r ve ilk ge�i� etkisinin minimal oldu�una i�aret etmektedir. Niraparibin pop�lasyon farmakokinetik analizinde, biyoyararlan�m a��s�ndan bireyler aras� de�i�kenli�in de�i�im katsay�s� (VK)
%31'dir.
300 mg niraparib uygulamas�ndan sonra, e�zamanl� y�ksek ya�l� ���n t�ketilmesi niraparibin farmakokineti�ini anlaml� d�zeyde etkilememi�tir.
Tablet ve kaps�l form�lasyonlar�n�n biyoe�de�er oldu�u g�sterilmi�tir. A�l�k ko�ullar� alt�nda kitle t�m�r� olan 108 hastada bir 300 mg tablet veya �� 100 mg niraparib kaps�l�n�n uygulanmas�n� takiben, tablet i�in Cmaks, EAAlast ve EAA∞ i�in kaps�llere k�yasla geometrik ortalama oranlar�n�n %90 g�ven aral�klar� biyoe�de�erlik limitlerinde olmu�tur (0,80 ve 1,25).
Da��l�m:
Niraparib insan plazmas�nda proteinlere orta d�zeyde (%83,0), esas olarak serum alb�minine
ba�lanmaktad�r. Niraparibin pop�lasyon farmakokinetik analizinde, kanser hastalar�nda (VK
%116) g�r�len da��l�m hacmi (V/F) 1,331 L'dir (70 kg hasta a��rl���na g�re) ve niraparibin
dokulara yayg�n olarak da��ld���na i�aret etmektedir. Biyotransformasyon:
Niraparib a��rl�kl� olarak karboksilesterazlar (CE) taraf�ndan metabolize edilir ve maj�r inaktif metabolit olan M1 olu�ur. Bir k�tle balans �al��mas�nda, M1 ve M10 (sonradan olu�turulan M1 glukuronidleri), dola��mdaki maj�r metabolitlerdir.
Eliminasyon:
Tek oral 300 mg niraparib dozundan sonra, niraparibin ortalama terminal yar� �mr� (t) 48 ila 51 saattir (yakla��k 2 g�n). Pop�lasyon farmakokinetik analizinde, niraparibin g�r�n�r toplam klirensi (CL/F) kanser hastalar�nda 16,5 L/h'dir (VK %23,4).
Niraparib esas olarak hepatobiliyer ve renal yollardan elimine edilmektedir. Tek 300 mg [C]- niraparib dozu uyguland�ktan sonra, dozun ortalama %86,2'si (aral�k %71 ila %91) 21 g�nl�k bir s�rede idrar ve d��k�da geri kazan�lm��t�r. �drardaki radyoaktivite geri kazan�m� dozun
%47,5'i (aral�k %33,4 ila %60,2) ve d��k�da %38,8'idir (aral�k: %28,3 ila %47). 6 g�nl�k bir s�rede toplanan havuzlanm�� numunelerde, dozun %40'� idrarda a��rl�kl� �ekilde metabolit olarak, dozun %31,6's� d��k�da esas olarak de�i�memi� niraparib olarak geri kazan�lm��t�r.
Hastalardaki karakteristik �zellikler B�brek yetmezli�i
Pop�lasyon farmakokinetik analizinde, hafif (kreatinin klirensi 60-90 mL/dk.) ve orta dereceli (30-60 ml/dk.) b�brek yetmezli�i olan hastalarda, b�brek i�levi normal (hafif yetmezlikte %7- 17 daha y�ksek maruziyet ve orta dereceli yetmezlikte %17-38 daha y�ksek maruziyet) olan bireylerle kar��la�t�r�ld���nda niraparib klirensi hafif�e d��m��t�r. Maruziyet fark�n�n doz ayarlamas� gerektirdi�i d���n�lmemektedir. Klinik �al��malarda �nceden �iddetli b�brek yetmezli�i veya son d�nem b�brek hastal��� olan ve hemodiyaliz g�ren hi�bir hasta belirlenmemi�tir (bkz. B�l�m 4.2).
Karaci�er yetmezli�i
Hastalarda yap�lan klinik �al��malarda elde edilen verilerin pop�lasyon farmakokinetik analizinde, �nceden var olan hafif dereceli karaci�er yetmezli�i (n=155) niraparibin klirensini etkilememi�tir. Karaci�er yetmezli�inin derecesini s�n�fland�rmak i�in NCI-ODWG kriterlerinin kullan�ld��� ve kanser hastalar�nda yap�lan bir klinik �al��mada, 300 mg'lik tek bir dozun uygulanmas�n�n ard�ndan orta dereceli karaci�er yetmezli�i bulunan hastalarda (n=8) niraparib EAA∞'s�, normal karaci�er fonksiyonu bulunan hastalardaki (n=9) niraparib EAA∞'s�n�n 1,56 kat� olmu�tur (%90 GA: 1,06 ila 2,30). Orta derecede karaci�er yetmezli�i olan hastalarda niraparib doz ayarlamas� gerekmektedir (bkz. B�l�m 4.2). Orta karaci�er yetmezli�inin niraparib C'� veya niraparib protein ba�lanmas� �zerinde bir etkisi olmam��t�r. Niraparibin farmakokineti�i �iddetli karaci�er yetmezli�i olan hastalarda de�erlendirilmemi�tir (bkz. B�l�m 4.2 ve B�l�m 4.4).
Kilo, ya� ve �rk
Pop�lasyon farmakokinetik analizinde a��rl�k art���n�n niraparibin da��l�m hacmini artt�rd��� g�r�lm��t�r. A��rl���n niraparib klirensi veya toplam maruziyet �zerinde hi�bir etkisi saptanmam��t�r. Farmakokinetik a��dan, v�cut a��rl���na g�re doz ayarlamas� gerekmemektedir.
Pop�lasyon farmakokinetik analizinde ya� art���n�n niraparib klirensini azaltt��� g�r�lm��t�r. 91 ya��ndaki bir hastadaki ortalama maruziyetin, 30 ya��ndaki bir hastadan %23 daha y�ksek olaca�� �ng�r�lm��t�r. Ya��n etkisinin doz ayarlamas� gerektirdi�i d���n�lmemektedir.
Irk�n niraparibin farmakokineti�i �zerindeki etkisiyle ilgili bir sonuca varmak i�in, �rklarda yeterli veri bulunmamaktad�r.
Pediyatrik pop�lasyon
Niraparibin pediyatrik hastalardaki farmakokineti�ini incelemek i�in hen�z �al��ma yap�lmam��t�r.
5.3. Klinik �ncesi g�venlilik verileri
G�venlilik farmakolojisi
�n vitro ortamda, niraparib insan maruziyet d�zeylerinin alt�ndaki konsantrasyonlarda dopamin ta��y�c�s� DAT'� inhibisyona u�ratm��t�r. Farelerde, tek doz niraparib, korteksteki h�cre i�i dopamin ve metabolit d�zeylerini artt�rm��t�r. Farelerde yap�lan iki tek doz �al��mas�ndan birinde lokomotor aktivitede d���� g�r�lm��t�r. Bu bulgular�n klinik �nemi bilinmemektedir. Beklenen terap�tik maruziyet d�zeylerine benzer veya bunlar�n alt�nda olmas� beklenen tahmini MSS Merkezi Sinir Sistemi (MSS) maruziyeti d�zeylerinde s��anlar ve k�peklerde yap�lan yinelenen dozlu toksisite �al��malar�nda davran��sal ve/veya n�rolojik parametreler �zerinde hi�bir etki g�zlemlenmemi�tir.
Yinelenen doz toksisitesi
Klinik olarak g�r�len maruziyet d�zeylerinde s��anlar ve k�peklerde spermatojenezde d���� g�zlenmi�tir ve dozun kesilmesini izleyen 4 hafta b�y�k oranda geri d�n��l�d�r.
Genotoksisite
Niraparib bakteriyel ters mutasyon tayininde (Ames) mutajenik de�ildir ama in vitro memeli
kromozom aberasyon analizinde ve in vivo s��an kemik ili�i mikron�kleus tayininde
klastojeniktir. Bu klastojenisite, niraparibin esas farmakolojisinden kaynaklanan genomik instabiliteyle tutarl�d�r ve insanlarda genotoksisite potansiyeli oldu�una i�aret etmektedir.
�reme toksikolojisi
Niraparible �reme ve geli�im toksisitesi �al��mas� yap�lmam��t�r. Karsinojenisite
Niraparible karsinojenisite �al��mas� yap�lmam��t�r.
6. FARMAS�T�K �ZELL�KLER
6.1. Yard�mc� maddelerin listesi
Tablet �ekirde�i Krospovidon
Laktoz monohidrat (s���r s�t�nden elde edilir)
Magnezyum stearat Mikrokristalin sel�loz Povidon
Silikon dioksit
Tablet kaplama
Polivinil alkol
Titanyum dioksit (E 171) Polietilen glikol
Talk
Ferrosoferrik oksit (E 172)
6.2. Ge�imsizlikler
Ge�erli de�ildir.
6.3. Raf �mr�
30 ay.
6.4. Saklamaya y�nelik �zel tedbirler
30°C'nin alt�ndaki oda s�cakl���nda, orijinal ambalaj�nda saklay�n�z.
6.5. Ambalaj�n niteli�i ve i�eri�i
84 × 1 ve 56 × 1 film kapl� tablet i�eren kutularda oPa/Al/PVC-Al/vinil/akrilik blisterler.
T�m ambalaj boyutlar� pazara sunulmam�� olabilir.
6.6. Be�eri t�bbi �r�nden arta kalan maddelerin imhas� ve di�er �zel �nlemler
Kullan�lmam�� olan �r�nler ya da at�k materyaller “T�bbi At�klar�n Kontrol� Y�netmeli�i” ve Ambalaj At�klar�n�n Kontrol� Y�netmeli�i”ne uygun olarak imha edilmelidir.
 Omurilik zedelenmeleri
Omurilik zedelenmesini takip eden birka� g�n i�inde, hi�kimse hasarin ne kadar olacagini tahmin edemez. Buradaki sorun, omuriligin herhangi bir zedelenmesinden hemen sonra, bir omurilik sokunun olusmasidir.
Omurilik zedelenmeleri
Omurilik zedelenmesini takip eden birka� g�n i�inde, hi�kimse hasarin ne kadar olacagini tahmin edemez. Buradaki sorun, omuriligin herhangi bir zedelenmesinden hemen sonra, bir omurilik sokunun olusmasidir. |
 HIV ve Aids
HIV, Human Immunodeficiency Virus’d�r (�nsanlarda Ba����kl�k Sistemini Bozan
Vir�sd�r). Bu vir�s AIDS hastal���na sebep olur.
HIV ve Aids
HIV, Human Immunodeficiency Virus’d�r (�nsanlarda Ba����kl�k Sistemini Bozan
Vir�sd�r). Bu vir�s AIDS hastal���na sebep olur. |
 |
Ruh ve Ak�l Sa�l���m�z� Geli�tirmek �yi ak�l ve ruh sa�l��� sahip olmaktan ziyade, yapt���n�z �eylerdir. Ak�l ve ruhsal olarak sa�l�kl� olmak i�in kendinize de�er vermeli ve kendinizi kabul etmelisiniz. |
 |
A��r� Alkol Kullan�m�, Alkolizm Alkol ba��ml�l���, alkol kullan�m� ve alkol sorunlar� aras�ndaki fark� a��klamak g��t�r. �rne�in, ge�mi�te alkol kullanm�� olan bir kimsenin mutlaka alkol ba��ml�s� olmas� gerekmez. |
 |
Kalp Krizi Kalbe giden kan ak��� durdu�unda kalp krizi meydana gelir. |
�LA� GENEL B�LG�LER�
Glaxo Smith Kline �la�lar� San.Ve Tic.A.�
| Sat�� Fiyat� | 119372.92 TL [ 3 Aug 2026 ] |
| �nceki Sat�� Fiyat� | 119372.92 TL [ 27 Jul 2026 ] |
| Original / Jenerik | Original �la� |
| Re�ete Durumu | Normal Re�eteli bir ila�d�r. |
| Barkodu | 8699522099559 |
| Etkin Madde | Niraparib |
| ATC Kodu | L01XK02 |
| Birim Miktar | 100 |
| Birim Cinsi | MG |
| Ambalaj Miktar� | 56 |
| Antineoplastik ve �mm�nomod�lat�r Ajanlar > Di�er Kanser �la�lar� |
| �thal ( ref. �lke : Avusturya ) ve Be�eri bir ila�d�r. |