TRUXIMA 100 mg/10 ml IV inf�zyonluk ��zelti haz�rlamak i�in konsantre (2 flakon) K�sa �r�n Bilgisi
{ Rituksimab }
1. BE�ER� TIBB� �R�N�N ADI
TRUXIMA 100 mg/10 mL iv inf�zyonluk ��zelti haz�rlamak i�in konsantre Steril
2. KAL�TAT�F VE KANT�TAT�F B�LE��M
Etkin madde
Her bir flakon 10 mL'lik ��zelti i�inde 100 mg rituximab i�erir. ��zeltinin her mL'sinde 10 mg rituximab bulunur.
Rituximab insan IgGl sabit b�lgeleri ve s�ras�yla de�i�ken m�rin hafif zincir ve a��r zincir i�eren bir glikozile imm�noglobulin sunan, genetik m�hendisli�i ile �retilen kimerik fare/insan monoklonal antikorudur. Antikor, memelilerin (�in hamster over h�cresi) h�cre s�spansiyon k�lt�r�nde �retilir ve spesifik viral inaktivasyon ve ��karma prosed�rlerini i�erecek �ekilde afinite kromotografisi ve iyon de�i�tirme ile safla�t�r�lan bir biyobenzer ila�t�r.
Yard�mc� maddeler
Tri-sodyum sitrat dihidrat : 7,36 mg/mL
3. FARMAS�T�K FORMU
�nf�zyon i�in konsantre ��zelti i�eren flakon. ��zelti berrak, renksiz bir s�v�d�r.
4. KL�N�K �ZELL�KLER
4.1. Terap�tik endikasyonlar
TRUXIMA yeti�kinlerde a�a��daki endikasyonlar i�in endikedir:
Hodgkin-d��� lenfoma (NHL)
TRUXIMA'n�n, daha �nce tedavi edilmemi� evre III-IVfolik�ler lenfomas� olan yeti�kin hastalar�n tedavisinde kemoterapi ile birlikte kullan�lmas� endikedir. TRUXIMA idame tedavisi, ind�ksiyon tedavisine yan�t veren yeti�kin folik�ler lenfoma hastalar�n�n tedavisinde endikedir.
TRUXIMA monoterapisi, kemoresistan veya kemoterapiden sonra ikinci ya da daha sonraki relapslar�nda olan evre III-IV folik�ler lenfomal� yeti�kin hastalar�n tedavisinde endikedir.
TRUXIMA'n�n, CD20 pozitif diff�z b�y�k B h�creli hodgkin-d��� lenfomas� olan yeti�kin
hastalar�n tedavisinde CHOP (siklofosfamid, doksorubisin, vinkristin, prednizolon) kemoterapisi ile birlikte kullan�lmas� endikedir. TRUXIMA'n�n ya� aral��� ≥6 ay ila <18 ya� olan pediyatrik hastalarda, daha �nceden tedavi edilmemi� ileri evre CD20 pozitif diff�z b�y�k B h�creli lenfoma (DLBCL), burkitt lenfoma (BL)/burkitt l�semi (olgun B-h�creli akut l�semi) (BAL) veya burkitt-benzeri lenfoma (BLL) tedavisinde kemoterapi ile birlikte kullan�lmas� endikedir.
Kronik lenfositik l�semi (KLL)
TRUXIMA, daha �nce tedavi edilmemi� ve relaps/refrakter (n�kseden/diren�li) KLL hastalar�n�n tedavisinde kemoterapi ile kombinasyon halinde endikedir. TRUXIMA dahil monoklonal antikorlar ile daha �nce tedavi edilmi� ya da daha �nceki kemoterapi ile kombine kullan�lan TRUXIMA tedavisine refrakter hastalarda etkililik ve g�venlilik ile ilgili s�n�rl� veri mevcuttur.
Daha fazla bilgi i�in B�l�m 5.1'e bak�n�z.
Gran�lomat�z polianjiitis (Wegener) (GPA) ve mikroskobik polianjiitis (MPA)
Siklofosfamide diren�li veya siklofosfamid tedavisi verilemeyen ciddi, aktif gran�lomat�z polianjiitis (GPA, Wegener gran�lomat�z� olarak da bilinir) ve mikroskobik polianjiitis (MPA) hastalar�n�n tedavisinde glukokortikoidlerle kombine olarak TRUXIMA kullan�l�r.
Pemfigus vulgaris (PV)
TRUXIMA, orta ila �iddetli pemphigus vulgarisi olan hastalar�n tedavisinde endikedir.
4.2. Pozoloji ve uygulama �ekli
TRUXIMA, acil uygulama i�in t�m res�sitasyon olanaklar�n�n bulundu�u bir ortamda deneyimli bir sa�l�k mesle�i mensubunun yak�n g�zetimi alt�nda uygulanmal�d�r (Bkz. B�l�m 4.4).
Premedikasyon ve profilaktik ila�lar
Her TRUXIMA inf�zyonundan �nce, analjezik/antipiretik (�rn. parasetamol) ve antihistaminik ila�tan (�rn. difenhidramin) olu�an bir premedikasyon her zaman yap�lmal�d�r.
Hodgkin-d��� lenfoma ve kronik lenfositik l�semili yeti�kin hastalarda, TRUXIMA glukortikoid i�eren kemoterapiyle kombinasyon halinde verilmiyorsa, glukokortikoidlerle premedikasyon g�z �n�nde bulundurulmal�d�r.
Hodgkin-d��� lenfomal� pediyatrik hastalara TRUXIMA inf�zyonunun ba�lang�c�ndan 30 ila 60 dakika �ncesinde parasetamol ve H1 antihistamin (=difenhidramin veya e�de�eri) ile premedikasyon uygulanmal�d�r. Ek olarak, prednizon Tablo 1'de belirtildi�i �ekilde uygulanmal�d�r.
KLL hastalar� i�in, t�m�r lizis sendromu (TLS) riskini azaltmak amac�yla tedavi ba�lang�c�ndan 48 saat �ncesinde yeterli hidrasyon ve �rikostatik uygulanmaya ba�lanmas� ile profilaksi �nerilmektedir. Lenfosit say�lar� > 25x10/L olan t�m KLL hastalar�nda akut inf�zyon reaksiyonlar� ve/veya sitokin sal�verilme sendromunun oran�n� ve ciddiyetini azaltmak amac�yla, TRUXIMA inf�zyonundan k�sa bir s�re �nce 100 mg i.v. prednizon/prednizolon uygulanmas� �nerilmektedir.
Gran�lamat�z polianjiitis (Wegener) veya mikroskobik polianjiitis veya pemfigus vulgaris hastalar�nda inf�zyonla ili�kili reaksiyonlar�n (IRR) s�kl���n� ve ciddiyetini azaltmak i�in her TRUXIMA inf�zyonundan 30 dakika �nce tamamlanacak �ekilde 100 mg i.v. metilprednizolon
uygulanmal�d�r.
Gran�lomat�z polianjiitis (Wegener) ve mikroskobik polianjiitis hastalar�nda TRUXIMA'n�n ilk inf�zyonundan �nce 1 ila 3 g�nl�k 1.000 mg i.v./g�n metilprednizolon �nerilmektedir (metilprednizolonun son dozu TRUXIMA'n�n ilk inf�zyonu ile ayn� g�nde verilebilir). Ard�ndan TRUXIMA tedavisinin 4 haftal�k ind�ksiyon k�r� s�ras�nda ve sonras�nda oral prednizon 1 mg/kg/g�n (en fazla 80 mg/g�n ve daha sonra klinik duruma g�re m�mk�n oldu�unca h�zl� bir �ekilde azalt�l�r) uygulanmal�d�r.
GPA ve MPA veya PV hastalar�na TRUXIMA tedavisi s�ras�nda ve sonras�nda yerel klinik uygulama k�lavuzlar�na uygun �ekilde Pn�mosistis jiroveci pn�moni (PCP) profilaksisi �nerilmektedir.
Pozoloji/uygulama s�kl��� ve s�resi:
Hodgkin-d��� lenfoma
Folik�ler Hodgkin-d��� lenfoma Kombinasyon tedavisi
�nceden tedavi edilmemi� veya relaps/refrakter (n�kseden/diren�li) folik�ler lenfoma hastalar�n�n ind�ksiyon tedavisinde, kemoterapi ile kombinasyon halinde �nerilen TRUXIMA dozu, her siklusta 375 mg/m v�cut y�zey alan� (BSA) olacak �ekilde en fazla 8 siklustur.
TRUXIMA, kemoterapinin glukokortikoid bile�eninin i.v. yolla verilmesinden sonra her bir kemoterapi siklusunun ilk g�n�nde uygulanmal�d�r.
�dame tedavisi
�nceden tedavi edilmemi� folik�ler lenfoma
�nd�ksiyon tedavisine yan�t vermi�, �nceden tedavi edilmemi�, folik�ler lenfomal� hastalar i�in idame tedavisi olarak kullan�lan TRUXIMA'n�n �nerilen dozu hastal�k progresyonuna kadar ya da en fazla 2 y�ll�k s�re (toplam 12 inf�zyon) boyunca (ind�ksiyon tedavisinin son dozundan 2 ay sonra ba�layarak) 2 ayda bir 375 mg/m v�cut y�zey alan�d�r.
Relaps/refrakter (n�kseden/diren�li) folik�ler lenfoma
�nd�ksiyon tedavisine yan�t vermi� relaps/refrakter folik�ler lenfomal� hastalar i�in idame tedavisi olarak kullan�lan TRUXIMA'n�n �nerilen dozu, hastal�k progresyonuna kadar veya en fazla 2 y�ll�k s�re (toplam 8 inf�zyon) boyunca (ind�ksiyon tedavisinin son dozundan 3 ay sonra ba�layarak) 3 ayda bir 375 mg/m v�cut y�zey alan�d�r.
Monoterapi
Relaps/Refrakter (N�kseden/Diren�li) folik�ler lenfoma
Evre III-IV folik�ler lenfoma olan, kemoterapiye diren�li veya kemoterapiden sonra ikinci kez veya daha fazla n�ks olu�an yeti�kin hastalar i�in ind�ksiyon tedavisi olarak kullan�lan TRUXIMA monoterapisinin �nerilen dozu: 4 hafta s�reyle haftada bir kere i.v. inf�zyon yoluyla verilen 375 mg/m v�cut y�zey alan�d�r.
N�ks eden/refrakter folik�ler lenfoma i�in TRUXIMA monoterapisi ile ge�mi� tedaviye yan�t veren hastalarda TRUXIMA monoterapisiyle yeniden tedavi i�in �nerilen doz: 4 hafta s�reyle haftada bir kere i.v. inf�zyon yoluyla verilen 375 mg/m v�cut y�zey alan�d�r (Bkz. B�l�m 5.1).
Yeti�kinlerde diff�z b�y�k B h�creli Hodgkin-d��� lenfoma
TRUXIMA, CHOP (siklofosfamid, doksorubisin, prednizolon ve vinkristin) kemoterapisi ile kombinasyon �eklinde kullan�lmal�d�r. �nerilen TRUXIMA dozu, her kemoterapi siklusunun
1. g�n�nde, 8 siklus i�in, CHOP rejiminin glukokortikoid bile�eni i.v. yoldan uyguland�ktan sonra verilmek �zere, 375 mg/m v�cut y�zey alan�d�r. CHOP rejiminin �teki bile�enleri, TRUXIMA uyguland�ktan sonra verilmelidir. Diff�z b�y�k B h�creli Hodgkin-d��� lenfomada di�er kemoterapilerle kombinasyon halinde TRUXIMA'n�n g�venlili�i ve etkilili�i belirlenmemi�tir.
Tedavi s�ras�nda doz ayarlamalar�
TRUXIMA dozunda herhangi bir azaltma �nerilmemektedir. TRUXIMA, kemoterapi ile kombine halde uyguland���nda, kemoterap�tik ila�lar i�in ge�erli standart doz azaltmalar� yap�lmal�d�r.
Kronik lenfositik l�semi (KLL)
Daha �nce tedavi edilmemi� ve relaps/refrakter hastalar i�in kemoterapiyle kombinasyon halinde �nerilen TRUXIMA dozu, toplam 6 siklus olmak �zere, ilk tedavi siklusunun 0. g�n�nde uygulanan 375 mg/m v�cut y�zey alan� ve sonras�ndaki her siklusun 1. g�n�nde uygulanan 500 mg/m v�cut y�zey alan�d�r. Kemoterapi, TRUXIMA inf�zyonundan sonra verilmelidir.
Gran�lomat�z polianjiitis (Wegener) (GPA) ve mikroskobik polianjiitis (MPA)
GPA ve MPA remisyon tedavisinin ind�ksiyonu i�in �nerilen TRUXIMA dozu, 4 hafta s�reyle haftada bir kere i.v. inf�zyon yoluyla verilen 375 mg/m v�cut y�zey alan�d�r.
Pemfigus vulgaris
TRUXIMA'n�n pemfigus vulgaris tedavisinde �nerilen dozu, azalt�larak kesilen glukokortikoid k�r� ile birlikte, i.v. inf�zyon olarak uygulanan 1.000 mg ve bunu takiben iki hafta sonra uygulanan ikinci bir 1.000 mg i.v. inf�zyondur.
�dame tedavisi
500 mg'l�k idame i.v. inf�zyon 12. ve 18. aylarda ve sonras�nda klinik de�erlendirme sonucunda gerek duyulmas� halinde her 6 ayda bir verilmelidir.
Relaps tedavisi
Relaps geli�mesi halinde, hastalar 1.000 mg i.v. alabilir. Hekim klinik de�erlendirme sonucunda hastan�n glukokortikoide tekrar ba�lamas�n� veya dozunun artt�r�lmas�n� d���nmelidir.
Devam inf�zyonlar�, bir �nceki inf�zyondan en az 16 hafta sonra yap�labilir.
Uygulama �ekli:
TRUXIMA sadece ona ayr�lm�� damar yoluyla, tek ba��na intraven�z inf�zyon olarak uygulanmal�d�r. TRUXIMA t�m res�sitasyon olanaklar�n�n eksiksiz olarak haz�r bulundu�u bir sa�l�k kurulu�unda ve uzman bir hekimin yak�n g�zetimi alt�nda uygulanmal�d�r.
Haz�rlanm�� inf�zyon ��zeltilerini i.v. pu�e veya bolus yoluyla uygulamay�n�z (Bkz. B�l�m 6.6).
Hastalar, sitokin sal�verilme sendromu ba�lamas� a��s�ndan yak�ndan izlenmelidir (Bkz. B�l�m 4.4). �zellikle �iddetli dispne, bronkospazm veya hipoksi �eklinde �iddetli reaksiyon belirtileri geli�en hastalarda inf�zyon derhal kesilmelidir. Hodgkin-d��� lenfomal� hastalar daha sonra, uygun laboratuvar testleri ile t�m�r lizis sendromu belirtileri a��s�ndan ve g���s r�ntgeni ile pulmoner infiltrasyon a��s�ndan de�erlendirilmelidir. Semptomlar tamamen d�zelene kadar ve laboratuvar de�erleriyle g���s r�ntgeni bulgular� normalle�ene dek t�m hastalarda inf�zyona tekrar ba�lanmamal�d�r. Ayr�ca inf�zyona ba�lang�� olarak daha �nce uygulanan�n en �ok yar�s� kadar bir h�zda yeniden ba�lanmal�d�r. Ayn� �iddetli advers reaksiyonlar ikinci kez meydana gelirse tedavinin durdurulmas� y�n�nde bir karar vaka baz�nda ciddi olarak d���n�lmelidir.
Hafif veya orta dereceli inf�zyonla ili�kili reaksiyonlar (IRR) (Bkz. B�l�m 4.8), genelde inf�zyon h�z�n�n azalt�lmas�na yan�t vermektedir. Semptomlar d�zeldikten sonra inf�zyon h�z� artt�r�labilir.
�lk inf�zyon
�nerilen ilk inf�zyon h�z� 50 mg/saattir; ilk 30 dakikadan sonra 30 dakikada bir 50 mg/saatlik art��larla h�z maksimum 400 mg/saate ��kar�labilir.
�zleyen �nf�zyonlar T�m endikasyonlar
Sonraki TRUXIMA inf�zyonlar�na 100 mg/saat h�z�yla ba�lanabilir ve daha sonra her 30 dakikada bir 100 mg/saatlik art��larla h�z maksimum 400 mg/saate ��kar�labilir.
Pediyatrik hastalar – Hodgkin-d��� Lenfoma �lk �nf�zyon
�nerilen ilk inf�zyon h�z� 0,5 mg/kg/saattir (en fazla 50 mg/saat); a��r� duyarl�l�k veya inf�zyon ile ba�lant�l� reaksiyonlar g�r�lmezse 30 dakikada bir 0,5 mg/kg/saatlik art��larla h�z maksimum 400 mg/saate ��kar�labilir.
�zleyen �nf�zyonlar
Sonraki TRUXIMA inf�zyonlar�na 1 mg/kg/saat h�z�yla ba�lanabilir (en fazla 50 mg/saat) ve daha sonra her 30 dakikada bir 1 mg/kg/saatlik art��larla h�z maksimum 400 mg/saate ��kar�labilir.
�zel pop�lasyonlara ili�kin ek bilgiler:
B�brek/ Karaci�er yetmezli�i:
B�brek/ karaci�er yetmezli�i olan hastalarda �zel bir kullan�m s�z konusu de�ildir.
Pediyatrik pop�lasyon:
Hodgkin-d��� Lenfoma
Ya�� ≥ 6 ay ila 18 ya� aras�nda olan daha �nceden tedavi edilmemi�, ileri evre CD20 pozitif DLBCL/BL/BAL/BLL'li pediyatrik hastalarda, TRUXIMA sistemik Lenfoma Malign B (LMB) kemoterapisi ile kombinasyon halinde kullan�lmal�d�r (Tablo 1 ve Tablo 2'ye bak�n�z). TRUXIMA'n�n �nerilen dozu i.v. inf�zyon yoluyla verilen 375 mg/m v�cut y�zey alan�d�r. TRUXIMA i�in v�cut y�zey alan� haricinde doz ayarlamas� gerekli de�ildir.
TRUXIMA'n�n g�venlili�i ve etkilili�i ya�� ≥ 6 ay ila 18 ya� aras�nda olan pediyatrik hastalarda daha �nceden tedavi edilmemi�, ileri evre CD20 pozitif DLBCL/BL/BAL/BLL endikasyonu d���ndaki endikasyonlarda ortaya konmam��t�r. 3 ya��n alt�ndaki hastalarda sadece s�n�rl� veri mevcuttur. Daha fazla bilgi i�in B�l�m 5.1'e bak�n�z.
TRUXIMA, CD20 pozitif diff�z b�y�k B h�creli lenfomas� olan 0 ila 6 ay aras�ndaki pediyatrik hastalarda kullan�lmamal�d�r (Bkz. B�l�m 5.1).
Tablo 1. Pediyatrik Hastalarda Non-Hodgkin Lenfoma i�in TRUXIMA Uygulamas�n�n Pozolojisi
Siklus | Tedavi G�n� | Uygulama Detaylar� | |
Prefaz (COP) | TRUXIMA verilmez | - | |
�nd�ksiyon k�r� 1 (COPDAM1) | -2. G�n (prefaz�n 6. G�n�ne tekab�l eder) 1. TRUXIMA inf�zyonu | uygulanmal�d�r. | |
TRUXIMA, ilk TRUXIMA inf�zyonundan 48 saat sonra verilecektir. | |||
�nd�ksiyon k�r� 2 (COPDAM2) | -2. G�n 3. TRUXIMA inf�zyonu | 2. ind�ksiyon prednizon uygulamas� verilmez. | k�r�nde, TRUXIMA zaman�nda |
1. G�n 4. TRUXIMA inf�zyonu | TRUXIMA, ���nc� TRUXIMA inf�zyonundan 48 saat sonra verilecektir. | ||
Konsolidasyon k�r� 1 (CYM/CYVE) | 1. G�n 5. TRUXIMA inf�zyonu | Prednizon uygulamas� verilmez. | TRUXIMA zaman�nda |
Konsolidasyon k�r� 2 (CYM/CYVE) | 1. G�n 6. TRUXIMA inf�zyonu | Prednizon uygulamas� verilmez. | TRUXIMA zaman�nda |
�dame k�r 1 (M1) | Konsolidasyon k�r� (CYVE) 25 ila 28. G�n� TRUXIMA verilmez. | 2'nin | Konsolidasyon k�r� 2'den (CYVE) sonra periferik say�lar d�zeldi�inde (ANC>1,0 x 10/L ve trombositler 100 x 10/L) ba�lar |
�dame k�r 2 (M2) | �dame k�r 1'n�n (M1) 28. G�n� TRUXIMA verilmez | - | |
ANC = Mutlak N�trofil Say�s�; COP = Siklofosfamid, Vinkristine, Prednizon; COPDAM = Siklofosfamid, Vinkristin, Prednizolon, Doksorubisin, Metotreksat; CYM = CYtarabin (Aracytine, Ara-C), Metotreksat; CYVE = CYtarabin (Aracytine, Ara-C), VEposid (VP16) | |||
ind�ksiyon k�r� esnas�nda, prednizon kemoterapi k�r�n�n bir par�as� olarak verilir ve TRUXIMA'dan �nce
4.3. Kontrendikasyonlar
TRUXIMA'n�n Hodgkin-d��� lenfoma ve kronik lenfositik l�semide kullan�m i�in kontrendike oldu�u durumlar:
Etkin madde
4.4. �zel kullan�m uyar�lar� ve �nlemleri

inf�zyon reaksiyonlar�n�n yakla��k %80'i ilk inf�zyonla ili�kili olarak g�r�lm��t�r. �nf�zyon s�ras�nda hastalar� dikkatlice g�zlemleyiniz. Evre 3 veya 4 inf�zyon reaksiyonlar� geli�irse TRUXIMA inf�zyonunu kesiniz ve t�bbi tedavi uygulay�n�z (Bkz. B�l�m 4.4, B�l�m 4.8).
T�m�r Lizis Sendromu (TLS)
Hodgkin-d��� lenfoma (NHL) hastalar�n�n TRUXIMA tedavisi sonras�nda, TLS sonucu, diyaliz gerektiren ve �l�mle sonu�lanan akut renal yetmezlik g�r�lebilir.
Ciddi Mukok�tan�z Reaksiyonlar
TRUXIMA kullanan hastalarda �l�mc�l olabilen, ciddi mukok�tan�z reaksiyonlar meydana gelebilir (Bkz. B�l�m 4.4, B�l�m 4.8).
Progresif Multifokal L�koensefalopati (PML)
TRUXIMA kullanan hastalarda PML ile sonu�lanan John Cunningham (JC) vir�s� aktivasyonu ve �l�m meydana gelebilir.
Hepatit B Vir�s (HBV) Reaktivasyonu
TRUXIMA ile tedavi edilen hastalarda fulminan hepatit, hepatik yetmezlik ve �l�mle sonu�lanabilen hepatit B reaktivasyonu ger�ekle�ebilir. TRUXIMA tedavisine ba�lamadan �nce b�t�n hastalar HBV enfeksiyonu a��s�ndan taranmal� ve tedavi s�resince ve sonras�nda hastalar izlenmelidir. HBV reaktivasyonu geli�en hastalarda acilen TRUXIMA ve birlikte kullan�lan kemoterapi ila�lar� kesilmelidir.
Progresif multifokal l�koensefalopati (PML)
Rituximab�n kullan�m� sonras�nda �ok seyrek �l�mc�l progresif multifokal l�koensefalopati (PML) vakalar� bildirilmi�tir. Hastalar, herhangi bir yeni veya k�t�le�en n�rolojik semptomlar a��s�ndan veya PML'yi d���nd�rebilecek belirtiler a��s�ndan d�zenli aral�klarla izlenmelidir. PML'den ��phelenilmesi durumunda, PML d��lanana kadar doz uygulamas� ask�ya al�nmal�d�r. Klinisyen, semptomlar�n n�rolojik disfonksiyonu g�sterip g�stermedi�ine ve n�rolojik disfonksiyon varsa, bu semptomlar�n PML'yi d���nd�r�p d���nd�rmedi�ine karar vermek i�in hastay� de�erlendirmelidir. Klinik gereklilik nedeniyle, bir n�rolog taraf�ndan kons�ltasyon yap�lmas� da dikkate al�nmal�d�r.
Herhangi bir ��phe olmas� durumunda tercihen kontrastl� MRG, JC Viral DNA i�in beyin- omurilik s�v�s� (BOS) testi ve tekrarl� n�rolojik de�erlendirmeler dahil detayl� muayene dikkate al�nmal�d�r.
Hekim, �zellikle hastan�n fark etmeyebilece�i PML semptomlar� (�rn. bili�sel, n�rolojik veya psikiyatrik semptomlar) a��s�ndan dikkatli olmal�d�r. Hastan�n fark�nda olmad��� semptomlar� fark edebilmeleri nedeniyle, hastalar�n ayr�ca e�leri ve hastaya bakanlar tedavi konusunda bilgilendirilmelidirler.
E�er bir hastada PML geli�irse, rituximab�n kullan�m� kal�c� �ekilde durdurulmal�d�r.
PML olan ba����kl��� zay�flam�� hastalarda imm�n sistemin yeniden d�zenlenmesi ard�ndan, stabilizasyon veya sonu�larda iyile�me oldu�u g�r�lm��t�r. PML'nin erken saptanmas�n�n ve rituximab tedavisinin ask�ya al�nmas�n�n benzer stabilizasyon veya sonu�larda iyile�me sa�lay�p sa�lamayaca�� bilinmemektedir.
Hodgkin-d��� lenfoma ve kronik lenfositik l�semi �nf�zyonla ili�kili reaksiyonlar
Rituximab sitokinlerin ve/veya di�er kimyasal arac�lar�n sal�nmas� ile ba�lant�l� olabilen
inf�zyon reaksiyonlar� ile ili�kilendirilmektedir. Sitokin sal�verilme sendromu klinik olarak akut a��r� duyarl�l�k reaksiyonlar�ndan ay�rt edilemeyebilir.
Sitokin sal�verilme sendromu, t�m�r lizis sendromu ve anafilaktik ve a��r� duyarl�l�k reaksiyonlar�n� i�eren reaksiyonlar toplulu�u a�a��da tarif edilmi�tir.
Rituximab�n intraven�z form�lasyonunun pazarlama sonras� deneyimlerde kullan�m� esnas�nda, ilk intraven�z rituximab inf�zyonunun ba�lamas�n�n ard�ndan 30 dakika ila 2 saat i�erisinde ba�layan ve �l�mle sonu�lanan ciddi inf�zyon reaksiyonlar� rapor edilmi�tir. Bunlar pulmoner olaylar ile karakterizedir ve baz� vakalarda ate�, titreme, kas�lma, hipotansiyon, �rtiker, anjiyo�dem ve di�er semptomlara ek olarak h�zl� t�m�r lizis ve t�m�r lizis sendromu �zelliklerini de i�ermi�tir (Bkz. B�l�m 4.8).
�iddetli sitokin sal�verilme sendromu ate�, titreme, kas�lma, �rtiker ve anjiyo�deme ek olarak s�kl�kla bronkospazm ve hipoksinin e�lik etti�i �iddetli dispne ile karakterizedir. Bu sendrom t�m�r lizis sendromunun hiper�risemi, hiperkalemi, hipokalemi, hiperfosfatemi, akut b�brek yetmezli�i, laktat dehidrojenaz (LDH) art��� gibi baz� �zellikleriyle ili�kili olabilir ve akut solunum yetmezli�ine ve �l�me yol a�abilir. Akut solunum yetmezli�ine, g���s r�ntgeninde g�r�nebilen pulmoner interstisyel infiltrasyon veya �dem gibi olaylar e�lik edebilir. Sendrom, genellikle ilk inf�zyonun ba�lat�lmas�ndan sonraki bir ya da iki saat i�inde kendini g�sterir. Ge�mi�te pulmoner yetmezli�i olan hastalar veya pulmoner t�m�r infiltrasyonu bulunan hastalar zay�f sonu�lar a��s�ndan daha fazla risk alt�nda olabilirler ve bu hastalar daha dikkatli tedavi edilmelidirler. �iddetli sitokin sal�verilme sendromu geli�en hastalar�n inf�zyonu derhal kesilmelidir (Bkz. B�l�m 4.2) ve bu hastalara agresif semptomatik tedavi uygulanmal�d�r. Klinik semptomlarda ba�ta g�r�len iyile�menin ard�ndan k�t�le�me olabilece�inden, bu hastalar t�m�r lizis sendromu ve pulmoner infiltrasyon ge�ene kadar veya d��lanana kadar yak�ndan izlenmelidir. Belirtilerin ve semptomlar�n tamamen ortadan kalkmas� ard�ndan hastalara uygulanan tedavi, nadiren �iddetli sitokin sal�verilme sendromunun tekrarlamas�yla sonu�lanm��t�r.
�zellikle �iddetli sitokin sal�verilme sendromu a��s�ndan y�ksek risk alt�nda olabilecek, KLL'si olan hastalar gibi y�ksek t�m�r y�k� veya dola��mda y�ksek say�da malign h�cresi (≥ 25 x 10/L) olan hastalar a��r� dikkatle tedavi edilmelidir. Bu hastalar ilk inf�zyonun ba��ndan sonuna kadar �ok yak�ndan g�zlemlenmelidir. Bu hastalarda, ilk inf�zyon s�ras�nda d���k bir inf�zyon h�z�n�n kullan�lmas� veya ilk siklus s�ras�nda ve lenfosit say�s�n�n hala >25 x 10/L olmas� durumunda takip eden sikluslarda dozun iki g�ne b�l�nerek verilmesi g�z �n�nde bulundurulmal�d�r.
�nf�zyonla ili�kili t�m advers reaksiyon tipleri, tedavi uygulanan hastalar�n %77'sinde g�zlenmi�tir (hastalar�n %10'unda hipotansiyon ve bronkospazm�n e�lik etti�i sitokin sal�verilme sendromu dahil, Bkz. B�l�m 4.8). Bu semptomlar genellikle rituximab inf�zyonunun kesilmesiyle ve bir antipiretik, bir antihistaminik ve baz� durumlarda oksijen, intraven�z serum fizyolojik veya bronkodilat�rlerin ve gerekti�inde glukokortikoidlerin uygulanmas�yla geri d�nd�r�lebilir olmu�tur. �iddetli reaksiyonlar i�in yukar�da yer alan sitokin sal�verilme sendromuna bak�n�z.
Hastalara intraven�z yolla protein verilmesinden sonra anafilaktik reaksiyonlar veya di�er a��r� duyarl�l�k reaksiyonlar� bildirilmi�tir. Sitokin sal�verilme sendromunun tersine, ger�ek a��r�
duyarl�l�k reaksiyonlar� tipik �ekilde inf�zyona ba�lanmas�ndan sonra dakikalar i�inde olu�ur. Rituximab uygulamas� s�ras�ndaki alerjik reaksiyon olgular�nda acil kullan�m i�in, a��r� duyarl�l�k reaksiyonlar�n�n tedavisine y�nelik ila�lar �rn., epinefrin (adrenalin), antihistaminikler ve glukokortikoidler kullan�ma haz�r bulundurulmal�d�r. Anafilaksinin klinik belirtileri, sitokin sal�verilme sendromunun klinik belirtilerine (yukar�da tan�mlanm��t�r) benzer g�r�nebilir. A��r� duyarl�l��a ba�l� reaksiyonlar, sitokin sal�verilmesine ba�l� reaksiyonlardan daha az s�kl�kta bildirilmi�tir.
Baz� vakalarda bildirilen di�er reaksiyonlar miyokard enfarkt�s�, atriyal fibrilasyon, pulmoner �dem ve akut geri d�nd�r�lebilir trombositopeni olmu�tur.
Rituximab inf�zyonu s�ras�nda hipotansiyon olu�abilece�inden, rituximab inf�zyonundan �nceki 12 saatlik s�re boyunca herhangi bir antihipertansif ilac�n al�nmam�� olmas�na dikkat edilmelidir.
Kardiyak hastal�klar
Rituximab ile tedavi edilen hastalarda anjina pektoris, atriyal flatter ve fibrilasyon gibi kardiyak aritmiler, kalp yetmezli�i ve/veya miyokard enfarkt�s� meydana gelmi�tir. Bu nedenle kardiyak hastal�k ve/veya kardiyotoksik kemoterapi �yk�s� olan hastalar yak�ndan izlenmelidir.
Hematolojik toksisiteler
Monoterapi �eklinde uygulanan rituximab miyelosupresif olmad��� halde, n�trofil say�s� < 1,5 x 10/L ve/veya trombosit say�s� < 75 x 10/L olan hastalar rituximab ile tedavi edilirken dikkatli olunmal�d�r, ��nk� bu tip hastalarla ilgili klinik deneyimler s�n�rl�d�r. Rituximab, otolog kemik ili�i transplantasyonu olan 21 hastada ve miyelotoksisite ind�klenmedi�i halde kemik ili�i fonksiyonlar�nda azalma olan di�er risk gruplar�nda kullan�lm��t�r.
Rituximab tedavisi s�ras�nda d�zenli olarak n�trofil ve trombosit say�m� dahil, kan h�crelerinin say�m� yap�lmal�d�r.
Enfeksiyonlar
Rituximab tedavisi s�ras�nda �l�mc�l, ciddi enfeksiyonlar meydana gelebilir (Bkz. B�l�m 4.8). Rituximab aktif, �iddetli enfeksiyonu (�rn. t�berk�loz, sepsis ve f�rsat�� enfeksiyonlar, Bkz. B�l�m 4.3) bulunan hastalara uygulanmamal�d�r.
Doktorlar, tekrarlayan veya kronik enfeksiyon �yk�s� bulunan veya hastay� ciddi enfeksiyonlara e�ilimli hale getirecek �ekilde altta yatan ko�ullara sahip hastalarda rituximab kullan�rken dikkatli olmal�d�rlar (Bkz. B�l�m 4.8).
Rituximab alan olgularda, �l�mle sonu�lanan fulminan hepatit de dahil olmak �zere hepatit B reaktivasyonu vakalar� bildirilmi�tir. Bu vakalar�n b�y�k �o�unlu�u ayr�ca sitotoksik kemoterapiye maruz kalm��t�r. N�kseden/refrakter KLL hastalar�nda yap�lan bir �al��madan sa�lanan k�s�tl� veriler, rituximab tedavisinin ayr�ca primer hepatit B enfeksiyonlar�n�n sonucunu k�t�le�tirebildi�ini g�stermektedir. Rituximab ile tedaviye ba�lanmadan �nce b�t�n hastalar (sadece HBV enfeksiyon riski olanlar de�il) Hepatit B vir�s� (HBV) a��s�ndan taranmal�d�r. Bu �l��mler en az�ndan hepatit B y�zey antijeni (HBsAg)-durumu ve hepatit B �ekirdek antikoru (HBcAb)-durumunu i�ermelidir. Bunlar yerel k�lavuzlara g�re di�er uygun mark�rler ile tamamlanmal�d�r. Aktif hepatit B hastal��� olan hastalar rituximab ile tedavi edilmemelidir. Pozitif hepatit B serolojisi olan hastalar (HBsAg veya HBcAb) tedavi ba�lang�c�ndan �nce karaci�er hastal�klar� uzman�na dan��mal�d�r ve bu hastalar hepatit B reaktivasyonunun �nlenmesi i�in yerel medikal standartlara g�re takip edilmeli ve y�netilmelidir.
NHL ve KLL'de rituximab�n pazarlama sonras� kullan�m� s�ras�nda �ok seyrek progresif multifokal l�koensefalopati (PML) vakalar� bildirilmi�tir (Bkz. B�l�m 4.8). Hastalar�n b�y�k �o�unlu�u rituximab� kemoterapi ile birlikte veya hematopoetik k�k h�cre transplantasyonunun bir par�as� olarak alm��lard�r.
�mm�nizasyonlar
NHL ve KLL hastalar�nda, rituximab tedavisini takiben canl� viral a��larla yap�lan imm�nizasyonun g�venlili�i �zerinde �al��ma yap�lmam��t�r ve canl� vir�s a��lar�yla a��lama yap�lmas� �nerilmemektedir. Rituximab ile tedavi edilen hastalar canl� olmayan a��larla a��lanabilirler. Ancak canl� olmayan a��lara yan�t oranlar� d��ebilir. Randomize olmayan bir �al��mada monoterapi olarak rituximab alan relaps, d���k evreli yeti�kin NHL hastalar� ile sa�l�kl�, tedavi g�rmemi� kontrol vakalar� kar��la�t�r�ld���nda, tetanoz hat�rlat�c� antijenine (%16'ya kar��l�k %81) ve Keyhole Limpet Haemocyanin (KLH) neoantijenine (antikor titrelerinde >2 kat� art��a g�re de�erlendirildi�inde %4'e kar��l�k %76) daha d���k oranda a�� yan�t� ger�ekle�mi�tir. Her iki hastal�k aras�ndaki benzerlikler dikkate al�nd���nda, KLL olan hastalar i�in benzer bulgular �ng�r�lebilir fakat klinik �al��malarda incelenmemi�tir.
Bir grup antijene kar�� (Streptococcus pneumoniae, influenza A, kabakulak, k�zam�k��k ve su�i�e�i) ortalama tedavi �ncesi antikor titreleri rituximab tedavisi sonras�nda en az 6 ay s�reyle korunmu�tur.
Deri reaksiyonlar�
Toksik Epidermal Nekroliz (Lyell sendromu) ve Stevens-Johnson sendromu gibi baz�lar�n�n �l�mc�l sonu�lar� olabilen ciddi cilt reaksiyonlar� bildirilmi�tir (Bkz. B�l�m 4.8). Bu gibi durumlarda olay�n rituximab ile ili�kili oldu�undan ��pheleniliyorsa tedavi kal�c� olarak durdurulmal�d�r.
Pediyatrik pop�lasyon
3 ya��n alt�ndaki hastalar i�in s�n�rl� veri mevcuttur. Daha fazla bilgi i�in B�l�m 5.1'e bak�n�z. Gran�lomat�z polianjiitis ve mikroskobik polianjiitis ve pemfigus vulgaris
�nf�zyonla ili�kili reaksiyonlar
Rituximab, sitokinlerin ve/veya di�er kimyasal mediyat�rlerin sal�verilmesine ba�l� olabilen inf�zyonla ilgili reaksiyonlarla (IRR) ili�kilendirilmi�tir.
Pazarlama sonras� ko�ullarda, romatoid artritli hastalarda �l�mc�l sonu�lara neden olabilen inf�zyonla ilgili �iddetli reaksiyonlar bildirilmi�tir. Klinik �al��malarda romatoid artrit hastalar�nda bildirilen inf�zyona ba�l� olgular�n �o�u hafif ve orta ciddiyette olmu�tur. En yayg�n semptomlar ba� a�r�s�, ka��nt�, bo�azda tahri�, k�zar�kl�k, d�k�nt�, �rtiker, hipertansiyon ve pireksidir. Genel olarak, herhangi bir tedavi k�r�n�n birinci inf�zyonunun ard�ndan herhangi bir inf�zyon reaksiyonu ya�ayan hastalar�n oran�, ikinci inf�zyonun ard�ndan g�r�lene oranla daha y�ksek olmu�tur. IRR insidans� sonraki k�rlerle azalm��t�r (Bkz. B�l�m 4.8). Bildirilen reaksiyonlar rituximab inf�zyonunun h�z�n�n azalt�lmas� ya da kesilmesine ve bir antipiretik, antihistaminik ve seyrek olarak oksijen, intraven�z serum fizyolojik veya bronkodilat�r�n ve gerekti�inde glukokortikoidlerin uygulanmas�na ba�l� olarak genellikle geri d�n���ml� olmu�tur. �nceden kalp sorunlar� bulunan ve �nceden kardiyopulmoner advers reaksiyon ya�am�� olan hastalar dikkatlice izlenmelidir. �nf�zyonla ilgili reaksiyonun ciddiyetine g�re ve gereken m�dahaleye g�re, rituximab kullan�m� ge�ici veya kal�c� olarak b�rak�lmal�d�r. Olgular�n �o�unda, semptomlar tamamen giderildi�inde, inf�zyon h�z� %50 oran�nda (�rn. 100 mg/saatten 50 mg/saat h�z�na) azalt�larak inf�zyona devam edilebilir.
Rituximab uygulamas� s�ras�ndaki alerjik reaksiyon olgular�nda acil kullan�m i�in, a��r� duyarl�l�k reaksiyonlar�n�n tedavisine y�nelik ila�lar �rn. epinefrin (adrenalin), antihistaminikler ve glukokortikoidler kullan�ma haz�r bulundurulmal�d�r.
Orta �iddette kalp yetmezli�i (NYHA s�n�f III) veya ciddi, kontrol alt�na al�nmam�� kardiyovask�ler hastal��� olan hastalarda rituximab kullan�m�yla ilgili g�venlilik verisi bulunmamaktad�r. Rituximab ile tedavi edilen hastalarda, atriyal fibrilasyon ve flatter ile anjina pektoris gibi �nceden mevcut olan iskemik kardiyak ko�ullar�n belirti verdi�i g�zlemlenmi�tir. Bu sebeple, rituximab tedavisinden �nce, bilinen bir kardiyak �yk�s� olan ve �nceden kardiyopulmoner advers reaksiyon ya�am�� olan hastalarda, inf�zyon reaksiyonlar�ndan kaynaklanan kardiyovask�ler komplikasyonlar�n riski dikkate al�nmal� ve hastalar uygulama s�ras�nda dikkatle g�zlenmelidir. Rituximab inf�zyonu s�ras�nda hipotansiyon olu�abilece�inden, rituximab inf�zyonu �ncesindeki 12 saatlik s�re boyunca herhangi bir antihipertansif ilac�n al�nmam�� olmas�na dikkat edilmelidir.
Gran�lomat�z polianjiitis ve mikroskobik polianjiitisi ve pemfigus vulgarisi bulunan hastalarda inf�zyonla ilgili reaksiyonlar, klinik �al��malarda romatoid artrit hastalar�nda g�r�lenlere benzer olmu�tur (Bkz. B�l�m 4.8).
Kardiyak hastal�klar
Rituximab ile tedavi edilen hastalarda anjina pektoris, atriyal flatter ve fibrilasyon gibi kardiyak aritmiler, kalp yetmezli�i ve/veya miyokard enfarkt�s� meydana gelmi�tir. Bu nedenle kardiyak hastal�k �yk�s� olan hastalar yak�ndan izlenmelidir (Bkz. yukar�da inf�zyonla ilgili reaksiyonlar).
Enfeksiyonlar
TRUXIMA'n�n etki mekanizmas� ve B h�crelerinin normal imm�n cevab�n s�rd�r�lmesinde �nemli rol oynamas� bilgisine dayanarak, rituximab tedavisi ard�ndan hastalar artan enfeksiyon riski ta��maktad�r (Bkz. B�l�m 5.1). Rituximab tedavisi s�ras�nda �l�mc�l, ciddi enfeksiyonlar meydana gelebilir (Bkz. B�l�m 4.8). Rituximab aktif, �iddetli enfeksiyonu (�rn. t�berk�loz, sepsis ve f�rsat�� enfeksiyonlar, Bkz. B�l�m 4.3) bulunan veya ba����kl��� ciddi d�zeyde bask�lanm�� (�rn. CD4 veya CD8 d�zeyleri �ok d���k olan) hastalara uygulanmamal�d�r. Doktorlar, tekrarlayan veya kronik enfeksiyon �yk�s� bulunan veya hastay� ciddi enfeksiyonlara e�ilimli hale getirecek �ekilde altta yatan ko�ullara (�rn. hipogamaglobulinemi) sahip hastalarda kullan�rken dikkatli olmal�d�rlar (Bkz. B�l�m 4.8). Rituximab tedavisine ba�lanmadan �nce imm�noglobulin d�zeylerinin saptanmas� �nerilir.
Rituximab tedavisi ard�ndan enfeksiyon belirti ve semptomlar� bildiren hastalar dikkatli �ekilde de�erlendirilmeli ve uygun �ekilde tedavi edilmelidir. Sonraki rituximab k�r� uygulanmadan �nce, hastalar potansiyel enfeksiyon riski a��s�ndan tekrar de�erlendirilmelidir.
Sistemik Lupus Eritematozus (SLE) ile vask�lit dahil otoimm�n hastal�klar�n tedavisi i�in rituximab kullan�m� ard�ndan �ok seyrek fatal progresif multifokal l�koensefalopati (PML) vakalar� bildirilmi�tir.
Hepatit B Enfeksiyonlar�
Rituximab alan gran�lomat�z polianjiitis ve mikroskobik polianjiitis hastalar�nda, �l�mle sonu�lananlar da dahil olmak �zere hepatit B reaktivasyonu vakalar� bildirilmi�tir.
Rituximab ile tedaviye ba�lanmadan �nce b�t�n hastalar (sadece HBV enfeksiyon riski olanlar de�il) Hepatit B vir�s� (HBV) a��s�ndan taranmal�d�r. Bu �l��mler en az�ndan hepatit B y�zey antijeni (HBsAg)-durumu ve hepatit B �ekirdek antikoru (HBcAb)-durumunu i�ermelidir. Bunlar yerel k�lavuzlara g�re di�er uygun mark�rler ile tamamlanmal�d�r. Aktif hepatit B hastal��� olan hastalar rituximab ile tedavi edilmemelidir. Pozitif hepatit B serolojisi olan hastalar (HBsAg veya HBcAb) tedavi ba�lang�c�ndan �nce karaci�er hastal�klar� uzman�na dan��mal�d�r ve bu hastalar hepatit B reaktivasyonunun �nlenmesi i�in yerel medikal standartlara g�re takip edilmeli ve y�netilmelidir.
Ge� n�tropeni
Rituximab ile tedaviye ba�lanmadan �nce ve tedavinin sonland�r�lmas� ard�ndan 6 aya kadar d�zenli olarak ve enfeksiyon belirti veya semptomlar�n�n g�r�lmesi durumunda, kan n�trofil �l��m� yap�lmal�d�r (Bkz. B�l�m 4.8).
Deri reaksiyonlar�
Toksik Epidermal Nekroliz (Lyell sendromu) ve Stevens-Johnson sendromu gibi baz�lar�n�n �l�mc�l sonu�lar� olabilen ciddi cilt reaksiyonlar� bildirilmi�tir (Bkz. B�l�m 4.8). Bu gibi durumlarda olay�n rituximab ile ili�kili oldu�undan ��pheleniliyorsa tedavi kal�c� olarak durdurulmal�d�r.
�mm�nizasyon
Hekimler, rituximab tedavisinden �nce hastan�n a��lanma durumunu de�erlendirmeli ve mevcut imm�nizasyon k�lavuzlar�n� izlemelidir. A��lama, ilk rituximab uygulamas�ndan en az 4 hafta �nce tamamlanm�� olmal�d�r.
Rituximab tedavisini takiben canl� viral a��larla yap�lan imm�nizasyonun g�venlili�i �zerinde �al��ma yap�lmam��t�r. Bu nedenle rituximab tedavisi s�ras�nda veya periferik B h�cre deplesyonu varken, canl� vir�s a��lar�yla a��lama yap�lmas� �nerilmemektedir.
Rituximab ile tedavi edilen hastalar canl� olmayan a��larla a��lanabilirler. Ancak canl� olmayan a��lara yan�t oranlar� d��ebilir. Randomize bir �al��mada, rituximab ve metotreksat ile tedavi edilen RA hastalar� ile yaln�zca metotreksat alan hastalar rituximab kullan�m�ndan en az 6 ay sonra a��land�klar�nda, tetanoz hat�rlat�c� antijenine kar�� benzer yan�t oran� (%39'a kar��l�k
%42), pn�mokokkal polisakkarid a��s�na (en az 2 pn�mokokkal antikor serotipine kar�� %43'e kar��l�k %82) kar�� ve KLH neoantijenine (%47'ye kar��l�k %93) kar�� ise azalm�� yan�t oran� g�stermi�lerdir. Rituximab tedavisi s�ras�nda canl� olmayan a��lama gerekli olursa, bunlar sonraki rituximab k�r�ne ba�lanmadan en az 4 hafta �nce tamamlanmal�d�r.
Malignite
�mm�nomod�lat�r ila�lar malignite riskini artt�rabilir. Mevcut veriler otoimm�n endikasyonlarda rituximab kullan�lmas� sebebiyle, halihaz�rda altta yatan otoimm�n hastal�kla ili�kili malignite riskinden daha fazla bir malignite riski oldu�unu d���nd�rmemektedir. Ancak solid t�m�r geli�imine ili�kin olas� risk g�z ard� edilemez.
Yard�mc� maddeler
TRUXIMA, flakon ba��na yakla��k 2,3 mmol (52,6 mg) sodyum ihtiva eder. Sodyum miktar� 1 mmol'den (23 mg) fazlad�r. Bu durum, kontroll� sodyum diyetinde olan hastalar i�in g�z �n�nde bulundurulmal�d�r.
Biyobenzer �r�nlerin takip edilebilirli�i
Biyobenzer t�bbi �r�nlerin takip edilebilirli�inin sa�lanmas� i�in uygulanan �r�n�n ticari ismi ve seri numaras� mutlaka hasta dosyas�na kaydedilmelidir.
4.5. Di�er t�bbi �r�nler ile etkile�imler ve di�er etkile�im �ekilleri
Rituximab ile olas� ila� etkile�imleri konusunda s�n�rl� veri vard�r.
KLL hastalar�nda, rituximab ile kombine kullan�m�n fludarabin veya siklofosfamidin farmakokineti�i �zerine bir etkisinin olmad���, bununla birlikte fludarabin veya siklofosfamidin de rituximab farmakokineti�i �zerine g�r�n�r bir etkisinin olmad��� g�r�lm��t�r.
�nsan anti-m�rin antikoru (HAMA) veya anti-ila� antikoru (ADA) titrelerine sahip hastalar tan� veya tedavi amac�yla ba�ka monoklonal antikorlarla tedavi edildiklerinde alerjik reaksiyonlar veya a��r� duyarl�l�k reaksiyonlar� geli�tirebilirler.
�zel pop�lasyonlara ili�kin ek bilgiler
Herhangi bir etkile�im �al��mas� yap�lmam��t�r.
4.6. Gebelik ve laktasyon
Gebelik kategorisi: C
�ocuk do�urma potansiyeli bulunan kad�nlar/Do�um kontrol� (kontrasepsiyon)
B h�cre deplesyonu olan hastalarda rituximab�n uzun retansiyon s�resi nedeniyle, �reme �a��ndaki kad�nlar rituximab tedavisi s�ras�nda ve bu tedaviyi takip eden 12 ay boyunca etkili do�um kontrol y�ntemleri kullanmal�d�r.
Gebelik d�nemi
Rituximab�n gebe kad�nlarda kullan�m�na ili�kin yeterli veri mevcut de�ildir. Hayvanlar �zerinde yap�lan �al��malar �reme toksisitesinin bulundu�unu g�stermi�tir (Bkz. B�l�m 5.3). �nsanlara y�nelik potansiyel risk bilinmemektedir.
IgG imm�noglobulinlerinin plasenta engelini ge�ti�i bilinmektedir.
Anne vas�tas�yla rituximaba maruz kalan insan yenido�an�ndaki B h�cre seviyeleri klinik �al��malarla ara�t�r�lmam��t�r. Gebe kad�nlarda yap�lm�� �al��malarda yeterli ve kontroll� veri elde edilememi�tir, ancak gebelik s�resince anneleri rituximaba maruz kalm�� olan baz� yenido�anlarda ge�ici B h�cre deplesyonu ve lenfositopeni bildirilmi�tir. Benzer etkiler hayvan �al��malar�nda da g�zlenmi�tir (Bkz. B�l�m 5.3). Bu sebeplerle rituximab, muhtemel faydalar potansiyel riskten fazla olmad��� s�rece gebe kad�nlarda uygulanmamal�d�r.
Laktasyon d�nemi
Rituximab�n anne s�t�yle at�lmas�na dair s�n�rl� veriler, anne s�t�nde �ok d���k seviyede (r�latif infant dozu %0,4'den az) oldu�unu d���nd�rmektedir. Takip edilen anne s�t�yle beslenen az say�daki bebek 1,5 ya��na kadar normal b�y�me ve geli�im g�stermi�tir. Ancak, bu veriler s�n�rl� oldu�u i�in ve bebeklerin emzirilmesinin uzun d�nem sonu�lar� bilinmedi�i i�in,
annelerin rituximab tedavisi s�ras�nda ve rituximab tedavisinden sonraki 12 ay boyunca bebeklerini emzirmemeleri tavsiye edilir.
�reme yetene�i/ Fertilite
Hayvan �al��malar�, rituximab�n �reme organlar� �zerinde zararl� etkileri oldu�unu g�stermemi�tir.
4.7. Ara� ve makine kullan�m� �zerindeki etkiler
Rituximab�n ara� veya makine kullanma becerisine etkisini belirleyecek �al��malar yap�lmam��t�r, ancak farmakolojik aktivite ve bug�ne kadar bildirilen yan etkiler rituximab�n ara� ve makine kullan�m� �zerinde hi�bir etkisinin olmad���n� ya da g�z ard� edilebilir etkilerinin oldu�unu d���nd�rmektedir.
4.8. �stenmeyen etkiler
Hodgkin-d��� lenfoma ve kronik lenfositik l�semi deneyimi G�venlilik profilinin �zeti
Hodgkin-d��� lenfoma ve kronik lenfositik l�semide rituximab�n genel g�venlilik profili, klinik �al��malarda ve pazarlama sonras� g�zetimde yer alan hastalardan gelen verilere dayanmaktad�r. Bu hastalar rituximab monoterapisiyle (ind�ksiyon tedavisi �eklinde veya ind�ksiyon tedavisini takiben idame tedavi �eklinde) veya kemoterapi ile kombinasyon halinde tedavi edilmi�tir.
Rituximab alan hastalarda en s�k g�zlenen advers ila� reaksiyonlar� (A�R'ler), hastalar�n �o�unlu�unda ilk inf�zyon s�ras�nda olu�an inf�zyonla ili�kili reaksiyonlar olmu�tur. �nf�zyonla ili�kili semptomlar�n insidans�, sonraki inf�zyonlarla belirgin �ekilde azalm��t�r ve sekiz doz rituximabdan sonra %1'den d���k olmu�tur.
Enfeksiy�z olaylar (a��rl�kl� �ekilde bakteriyel ve viral), yap�lan klinik �al��malar s�ras�nda NHL olan hastalarda yakla��k hastalar�n %30-55'inde ve KLL olan hastalarda hastalar�n %30- 50'sinde meydana gelmi�tir.
En s�k bildirilen veya g�zlenen ciddi advers ila� reaksiyonlar�:
�nf�zyonla ili�kili reaksiyonlar (IRR) (sitokin sal�verilme sendromu, t�m�r lizis sendromu dahil), Bkz. B�l�m 4.4
4.9. Doz a��m� ve tedavisi
�ntraven�z rituximab form�lasyonunun onaylanm�� dozundan daha y�ksek dozlarla ilgili olarak insanlarda yap�lan klinik �al��malarda deneyim k�s�tl�d�r. Bug�ne kadar insanlarda test edilen
en y�ksek intraven�z rutiximab dozu kronik lenfositik l�semi (KLL) hastalar� ile y�r�t�len doz art�rma �al��mas�nda test edilen 5.000 mg'd�r (2.250 mg/m). Hi�bir ek g�venlilik sinyali tespit edilmemi�tir.
Doz a��m� g�r�len hastalarda inf�zyon derhal kesilmelidir ve hastalar yak�ndan izlenmelidir.
Pazarlama sonras� ko�ullarda rituximab doz a��m�na ili�kin be� vaka bildirilmi�tir. �� vaka advers olay rapor etmemi�tir. Bildirilen iki advers olay, 1,8 g'l�k rituximab dozuyla raporlanan grip benzeri semptomlar ve 2 g'l�k rituximab dozuyla raporlanan �l�mc�l solunum yetmezli�idir.
5. FARMAKOLOJ�K �ZELL�KLER
5.1. Farmakodinamik �zellikler
Farmakoterap�tik grubu: Antineoplastik ajanlar, monoklonal antikorlar ATC kodu: L01FA01
Bu �r�n bir biyobenzer ila�t�r. Etki mekanizmas�
Rituximab spesifik olarak bir non-glikolize fosfoprotein olan CD20 adl� transmembran
antijenine ba�lanan kimerik fare/insan monoklonal antikorudur. Bu antijen pre-B ve olgun B lenfositlerinde bulunur. T�m B h�creli Hodgkin-d��� lenfomalar�n (NHL) >%95'inde bu antijen eksprese edilir.
CD20 hem normal hem de malignan B h�crelerinde bulunur ancak hematopoetik k�k h�crelerde, pro-B h�crelerde, normal plazma h�crelerinde veya di�er normal dokuda bulunmaz. Antikora ba�land�ktan sonra CD20 h�cre i�ine al�nmaz veya h�cre membran�ndan �evreye yay�lmaz. CD20 plazmada serbest antijen olarak dola�maz ve dolay�s�yla antikora ba�lanmak i�in yar��maz.
Rituximab�n Fab alan�, B lenfositlerdeki CD20 antijenine ba�lan�r ve Fc alan� B h�cresinin lizisine yol a�an ba����kl��� etkileyen i�levleri ba�lat�r. Etki-arac�l� h�cre y�k�m�ndaki olas� mekanizmalar C1q ba�lanmas� sonucunda olu�an komplemana ba��ml� sitotoksisite (CDC), gran�lositlerin, makrofajlar�n ve NK h�crelerinin y�zeyindeki Fcγ resept�rlerinin bir veya daha fazlas� arac�l���yla olu�an antikora ba��ml� h�cresel sitotoksisite (ADCC)'dir. B lenfositleri �zerindeki CD20 antijenine rituximab ba�lanmas�n�n apoptoz yoluyla h�cre �l�m�n� ind�kledi�i ortaya konmu�tur.
Periferdeki B h�cre say�s� ilk rituximab dozundan sonra normalin alt�na d��er. Hematolojik malignite tedavisi g�ren hastalarda B h�crelerinin normal d�zeyine d�nmesi tedavinin 6. Ay� i�inde ba�lar, baz� hastalarda daha uzun s�rse bile, genellikle tedavi tamamland�ktan sonraki 12 ay i�inde de normal d�zeylere d�ner (ind�ksiyon tedavisinden sonra medyan d�zelme s�resi 23 aya kadar uzayabilir). Romatoid artrit hastalar�nda, periferik kandaki B h�creleri say�s�n�n h�zla azalmas� 14 g�nl�k arayla verilen 1.000 mg'l�k iki rituximab inf�zyonundan sonra g�zlenmi�tir. Rituximab monoterapi veya metotreksat ile beraber kombine olarak uyguland���nda periferik kan B h�crelerinin say�s� 24. Haftadan sonra artmaya ba�lar ve hastalar�n b�y�k �o�unlu�unda 40. Haftada eski say�ya d�nd���n� g�steren i�aretler g�zlenmi�tir. Hastalar�n k���k bir k�sm�nda son rituximab dozundan sonraki 2 y�la kadar veya daha uzun s�re devam eden periferik B h�cre say�s� azalmas� olmu�tur.
Gran�lomat�z polianjiitis ve mikroskobik polianjiitis hastalar�nda, iki haftal�k 375 mg/m rituximab inf�zyonu sonras�nda periferik kan B h�creleri 10 h�cre/µL'nin alt�na d��m��t�r ve 6 ay boyunca hastalar�n �o�unda ayn� seviyede kalm��t�r. Hastalar�n b�y�k �o�unlu�u (%81)
12. Ayda B h�crelerinin 10 h�cre/µL seviyesinin �zerinde say�larla tekrar artmaya ba�lad���na dair i�aretler g�stermi�tir, bu oran 18. Ay itibariyle hastalar�n %87'sine y�kselmi�tir.
Hodgkin-d��� lenfoma ve kronik lenfosittik l�semide klinik deneyim Folik�ler lenfoma
Monoterapi
Ba�lang�� tedavisi, haftada bir uygulama, toplam 4 doz
Pivotal �al��mada, n�ksetmi� veya kemorezistan d���k seviyeli ya da folik�ler B h�creli NHL'ye sahip 166 hastaya haftada bir, toplam d�rt doz olarak i.v. inf�zyon halinde 375 mg/m rituximab verilmi�tir. Tedavi meyilli (ITT) pop�lasyonunda genel yan�t oran� (GYO) %48 (GA%41 - %56) olup tam yan�t (TY) oran� %6 ve k�smi yan�t (KY) oran� %42 olarak ger�ekle�mi�tir. Yan�t veren hastalarda hastal���n ilerlemesine kadar ge�en medyan s�re (TTP) 13 ay'd�r. Yap�lan bir alt-grup analizinde GYO, IWF B, C ve D histolojik alt-tiplerinde IWF A alt-tipine k�yasla daha y�ksek (%58'e kar��l�k %12), en b�y�k lezyonu <5 cm olan hastalarda,
>7 cm olan hastalara k�yasla daha y�ksek (%53'e kar��l�k %38) ve kemorezistan n�ks grubu ile kar��la�t�r�ld���nda kemoduyarl� n�ks hastalar�nda (yan�t s�resi <3 ay olarak tan�mlan�r) daha y�ksek (%22'ye kar��l�k %50) bulunmu�tur. �nceden otolog kemik ili�i transplantasyonu (OK�T) ile tedavi edilmi� hastalarda GYO %78 iken, OK�T tedavisi g�rmeyenlerde %43 olmu�tur. Ya�, cinsiyet, lenfoma derecesi, ba�lang��taki tan�, b�y�k hacimli hastal�k varl��� ya da yoklu�u, normal ya da y�ksek LDH d�zeyleri veya ekstranodal hastal�k varl���n�n rituximaba kar�� verilen yan�t �zerinde istatistik olarak anlaml�l�k (Fischer'in exact testi) ta��yan bir etkisi bulunmamaktad�r. Yan�t oranlar� ile kemik ili�i tutulumu aras�nda istatistiksel olarak anlaml� bir ba��nt� kaydedilmi�tir. Kemik ili�i tutulumu olan hastalar�n %40'� yan�t verirken, kemik ili�i tutulumu olmayan hastalar�n %59'u yan�t vermi�tir (p=0,0186). Histolojik tip, ba�lang��ta bcl-2 pozitifli�i, son kemoterapiye kar�� diren� ve b�y�k hacimli hastal�k fakt�rlerinin prognostik fakt�rler olarak tan�mland��� kademeli lojistik regresyon analizi bu bulguyu desteklememi�tir.
Ba�lang�� tedavisi, haftada bir uygulama, toplam 8 doz
�ok merkezli, tek kollu bir �al��mada n�ksetmi� veya kemorezistan, d���k dereceli veya folik�ler B h�creli NHL'si olan 37 hastaya, toplam sekiz doz olacak �ekilde, i.v. inf�zyon yoluyla haftada bir kere 375 mg/m rituximab verilmi�tir. GYO %57 (GA%41- %73; TY
%14, KY %43) ve medyan TTP 19,4 ay (5,3 ila 38,9 ay aral���nda) bulunmu�tur. Ba�lang�� tedavisi, b�y�k hacimli hastal�k, haftada bir uygulama, toplam 4 doz
�� �al��man�n bir araya getirilmi� verilerinde, n�ksetmi� veya kemorezistan, b�y�k hacimli
hastal�k (tek lezyon ≥ 10 cm �ap�nda) �zelliklerini ta��yan d���k dereceli veya folik�ler B h�creli NHL'si olan 39 hastaya toplam d�rt doz, haftada bir kere 375 mg/m rituximab, i.v. inf�zyon yoluyla verilmi�tir. GYO %36 (GA%21-%51; TY %3, KY %33) ve yan�t veren hastalarda medyan TTP 9,6 ay olmu�tur (4,5 ila 26,8 ay aral���nda).
Tekrarlanan tedavi, haftada bir uygulama, toplam 4 doz
�ok merkezli, tek kollu bir �al��mada, �nceki bir rituximab k�r�nde objektif klinik yan�t al�nan, n�ksetmi� veya kemorezistan d���k evreli veya folik�ler B h�creli NHL'si olan 58 hasta toplam d�rt doz, haftada bir kere, i.v. inf�zyon yoluyla 375 mg/m rituximab ile yeniden tedavi edilmi�tir. Hastalar�n ��� �al��maya kaydolmadan �nce iki seans rituximab tedavisi g�rd���nden, bunlara �al��mada ���nc� bir seans uygulanm��t�r. �al��mada iki hasta, iki kez yeniden tedavi edilmi�tir. �al��madaki 60 yeniden tedavi vakas�nda, yan�t veren hastalar i�in GYO %38 (GA%26-%51; TY %10, KY %28) ve yan�t veren hastalarda projekte edilen ortalama TTP, 17,8 ay (5,4 ila 26,6 ay aral���nda) olmu�tur. Bu de�erler, �nceki rituximab k�r�nde elde edilen sonu�lar (12,4 ay) ile olumlu y�nde kar��la�t�r�labilir niteliktedir.
Ba�lang�� tedavisi, kemoterapi ile kombinasyon halinde
Randomize, a��k etiketli bir �al��mada, daha �nce tedavi g�rmemi� folik�ler lenfomas� olan 322 hasta, 8 k�r, her 3 haftada bir CVP kemoterapisi (siklofosfamid 750 mg/m, 1. G�n maksimum 2 mg doza kadar, vinkristin 1,4 mg/m ve 1-5. G�n aras�nda prednizolon 40 mg/m/g�n) ya da CVP ile kombinasyon halinde rituximab 375 mg/m (R-CVP) alacak �ekilde randomize edilmi�tir. Rituximab her tedavi siklusunun ilk g�n�nde uygulanm��t�r. Toplam 321 hasta (162 R-CVP, 159 CVP) tedavi g�rm�� ve etkililik bak�m�ndan analiz edilmi�tir. Hastalar�n medyan takip s�resi 53 ayd�r. R-CVP, primer sonlanma noktas� olan tedavi ba�ar�s�zl���na kadar ge�en s�re a��s�ndan CVP'ye g�re �nemli bir �st�nl�k sa�lam��t�r (27 aya kar��l�k 6,6 ay, p < 0,0001, log-s�ra testi). T�m�r cevab� bulunan hastalar�n oran� (TY, TYo, KY), R-CVP grubunda (%80,9) CVP grubundan (%57,2) �nemli oranda daha y�ksek olmu�tur (p< 0,0001, Ki-Kare testi). R-CVP ile yap�lan tedavi, hastal�k ilerlemesi veya �l�me kadar ge�en s�reyi belirgin bir �ekilde art�rm��t�r (33,6 ay ve l4,7 ay) (p < 0,0001, log-s�ra testi). R- CVP grubunda medyan yan�t s�resi 37,7 ayken, CVP grubunda bu s�re 13,5 ay olarak bulunmu�tur (p < 0,0001, log-s�ra testi).
Genel sa�kal�m a��s�ndan tedavi gruplar� aras�ndaki farkl�l�k g��l� bir klinik yarar g�stermi�tir (p=0,029, katmanl� log-s�ra testi): 53. Aydaki sa�kal�m oranlar� R-CVP grubunda %80,9 iken, CVP grubunda %71,1 'dir.
CVP d���ndaki kemoterapi rejimleriyle (CHOP, MCP, CHVP/Interferon-α) yap�lan di�er 3 randomize �al��madan elde edilen sonu�lar yan�t oranlar�nda ve zamana ba�l� parametrelerde oldu�u gibi genel sa�kal�mda da belirgin iyile�meler g�stermi�tir. Bu d�rt �al��madan elde edilen anahtar sonu�lar a�a��da Tablo 6'da �zetlenmektedir.
Tablo 6: Rituximab�n folik�ler lenfomada farkl� kemoterapi rejimleriyle yararlar�n�n de�erlendirildi�i d�rt faz III randomize �al��madan elde edilen anahtar sonu�lar�n �zeti
�al��ma | Tedavi, N | Medyan Takip S�resi, ay |
GYO, % |
TY, % | Medyan TTF/PFS/ EFS ay | OS oranlar�, % |
M39021 |
CVP, 159 R-CVP, 162 |
53 |
57 81 |
10 41 | Medyan TTP: 14,7 33,6 p<0,0001 | 53 ay 71,1 80,9 p=0,029 |
GLSG'00 |
CHOP, 205 R-CHOP, 223 |
18
|
90 96 |
17 20 | Medyan TTF: 2,6 y�l Ula��lamam��t�r p< 0,001 | 18 ay 90 95 p = 0,016 |
OSHO-39 |
MCP, 96
R-MCP, 105 |
47 |
75
92 |
25 50 | Medyan PFS: 28,8 Ula��lamam��t�r p< 0,0001 | 48 ay 74 87 p = 0,0096 |
FL2000 | CHVP-IFN, 183
R-CHVP-IFN, 175 |
42 | 85
94 | 49 76 | Medyan EFS: 36 Ula��lamam��t�r p<0,0001 | 42 ay 84 91 p=0,029 |
GYO: Genel yan�t oran� TY: Tam yan�t
EFS: Olays�z sa�kal�m
TTP: Progresyona veya �l�me kadar ge�en s�re PFS: Progresyonsuz sa�kal�m
TTF: Tedavinin ba�ar�s�zl���na kadar ge�en s�re
OS oranlar�: Analizler zaman�nda genel sa�kal�m oranlar�
�dame tedavisi
Daha �nce tedavi uygulanmam�� folik�ler lenfoma
Prospektif, a��k etiketli, uluslararas�, �ok merkezli bir faz III �al��mada daha �nce tedavi uygulanmam�� ileri seviye folik�ler lenfomas� olan 1.193 hastaya, ara�t�rmac�n�n tercihine g�re R-CHOP (n=881), R-CVP (n=268) veya R-FCM (n=44) ile ind�ksiyon tedavisi uygulanm��t�r. Toplam 1.078 hasta ind�ksiyon tedavisine yan�t vermi�, bu hastalardan 1.018'i rituximab idame tedavisine (n=505) veya g�zlem grubuna (n=513) randomize edilmi�tir. �ki tedavi grubu, ba�lang��taki �zellikler ve hastal�k durumu a��s�ndan iyi dengelenmi�tir. Rituximab idame tedavisi, hastal�k progresyonuna kadar veya maksimum iki y�l boyunca 2 ayda bir 375 mg/m v�cut y�zey alan� dozunda tek rituximab inf�zyonundan olu�mu�tur.
Randomizasyondan itibaren 25 ayl�k medyan g�zlem s�resinde yap�lan, �nceden belirlenmi� bir primer analiz s�ras�nda rituximab idame tedavisi, �nceden tedavi edilmemi� folik�ler lenfoma hastalar�nda g�zlem grubuna k�yasla, ara�t�rmac� taraf�ndan de�erlendirilen progresyonsuz sa�kal�m (PFS) primer sonlan�m noktas�nda klinik olarak tedaviyle alakal� ve istatistiksel olarak anlaml� iyile�me sa�lam��t�r (Tablo 7).
Primer analizde rituximab ile idame tedaviden elde edilen anlaml� yarar ayr�ca sekonder
sonlan�m noktalar� olan olays�z sa�kal�m (EFS), bir sonraki anti-lenfoma tedavisine kadar ge�en s�re (TNLT), bir sonraki kemoterapiye kadar ge�en s�re (TNCT) ve genel yan�t oran�nda (GYO) da g�r�lm��t�r (Tablo 7).
�al��madaki hastalar�n uzun s�reli takibinden (medyan takip 9 y�l) elde edilen veriler rituximab�n idame tedavisinin PFS, EFS, TNLT ve TNCT a��s�ndan uzun s�reli yarar�n� teyit etmi�tir (Tablo 7).
Tablo 7: Protokolde tan�mlanm�� primer analizlerde ve 9 y�ll�k medyan takip (sonu� analizi) sonras�nda rituximab�n idame tedavisinin etkililik sonu�lar�n�n g�zlem grubu ile kar��la�t�r�lmas�n�n genel �zeti
| Primer analiz (medyan takip: 25 ay) | Son analiz (medyan takip: 9 y�l) | ||
| G�zlem N=513 | Rituximab N=505 | G�zlem N=513 | Rituximab N=505 |
Primer etkililik | ||||
Progresyonsuz sa�kal�m | NR NR | 4,06 y�l 10,49 y�l | ||
(medyan) |
|
| ||
log-s�ra p de�eri | <0,0001 | <0,0001 | ||
risk oran� (GA) | 0,50 (0,39, 0,64) | 0,61 (0,52, 0,73) | ||
risk azalmas� | %50 | %39 | ||
Sekonder etkililik | ||||
Genel sa�kal�m | NR NR | NR NR | ||
(medyan) |
|
| ||
log-s�ra p de�eri | 0,7246 | 0,7948 | ||
risk oran� (GA) | 0,89 (0,45, 1,74) | 1,04 (0,77, 1,40) | ||
risk azalmas� | %11 | %-6 | ||
Olays�z sa�kal�m | 38 ay NR | 4,04 y�l 9,25 y�l | ||
(medyan) |
|
| ||
log-s�ra p de�eri | <0,0001 | <0,0001 | ||
risk oran� (GA) | 0,54 (0,43, 0,69) | 0,64 (0,54, 0,76) | ||
risk azalmas� | %46 | %36 | ||
TNLT (medyan) | NR NR | 6,11 y�l NR | ||
log-s�ra p de�eri | 0,0003 | <0,0001 | ||
risk oran� (GA) | 0,61 (0,46, 0,80) | 0,66 (0,55, 0,78) | ||
risk azalmas� | %39 | %34 | ||
TNCT (medyan) | NR NR | 9,32 y�l NR | ||
log-s�ra p de�eri | 0,0011 | 0,0004 | ||
risk oran� (GA) | 0,60 (0,44, 0,82) | 0,71 (0,59, 0,86) | ||
risk azalmas� | %40 | %39 | ||
Genel Yan�t Oran�* | %55 %74 | %61 %79 | ||
Ki-kare testi p de�eri | <0,0001 | <0,0001 | ||
Olas�l�k oran� (GA) | 2,33 (1,73, 3,15) | 2,43 (1,84, 3,22) | ||
Tam yan�t (TY/TYo) | %48 %67 | %53 %67 | ||
oran�* |
|
| ||
Ki-kare testi p de�eri | <0,0001 | <0,0001 | ||
Olas�l�k oran� (GA) | 2,21 (1,65, 2,94) | 2,34 (1,80, 3,03) | ||
*�dame/g�zlem sonunda; son analiz sonu�lar� 73 ayl�k medyan takip s�resine dayanmaktad�r. GA: g�ven aral���; NR: klinik kesme tarihinde elde edilmemi�; TNCT: sonraki kemoterapiye kadar ge�en s�re; TNLT: sonraki anti-lenfoma tedavisine kadar ge�en s�re.
Rituximab idame tedavisi, �nceden tan�mlanm�� t�m test edilen alt gruplarda tutarl� yarar sa�lam��t�r: cinsiyet (erkek, kad�n), ya� (<60, ≥60), FLIPI skoru (≤1, 2 veya ≥3), ind�ksiyon tedavisi (R-CHOP, R-CVP veya R-FCM) ve ind�ksiyon tedavisine verilen yan�t�n niteli�inden (TY/TYo veya KY) ba��ms�zd�r. �dame tedavinin yarar�na ili�kin eksplatuar analizler, ya�l� hastalarda (>70 ya�) etkinin daha az belirgin oldu�unu g�stermi�tir ancak �rneklem say�s� azd�r.
Relaps/refrakter folik�ler lenfoma
Prospektif, a��k etiketli, uluslararas�, �ok merkezli bir faz III �al��mada 465 relaps/refrakter folik�ler lenfoma hastas�, CHOP (siklofosfamid, doksorubisin, vinkristin, prednizolon; n=231) veya rituximab + CHOP (R-CHOP, n=234) ile yap�lan ind�ksiyon tedavisine ilk basamakta randomize edilmi�tir. �ki tedavi grubu, ba�lang�� karakteristiklerine ve hastal�k durumuna g�re iyi dengelenmi�tir. �nd�ksiyon tedavisinden sonra tam ya da k�smi remisyon sa�lanan toplam 334 hasta, ikinci a�amada rituximab idame tedavisi (n=167) veya g�zlem koluna (n= 167) randomize edilmi�tir. Rituximab idame tedavisi, maksimum iki sene s�resince ya da hastal�k ilerleyene kadar, �� ayda bir 375 mg/m v�cut y�zey alan� dozunda verilen tek rituximab uygulamas�ndan ibarettir.
Son etkililik analizi, �al��man�n her iki b�l�m�ne randomize edilen t�m hastalar� i�erir. �nd�ksiyon faz�na randomize edilen hastalar�n 31 ayl�k medyan g�zlem s�resi sonunda, R- CHOP'un, CHOP ile k�yasland���nda relaps/refrakter folik�ler lenfoma hastalar�n�n klinik sonu�lar�n� belirgin olarak iyile�tirdi�i g�r�lm��t�r (Bkz. Tablo 8).
Tablo 8: �nd�ksiyon faz�: CHOP ile R-CHOP'un kar��la�t�rmal� etkililik sonu�lar�na genel bak�� (31 ayl�k medyan g�zlem s�resi)
| CHOP | R-CHOP | p de�eri | Risk Azalt�m� |
Primer Etkililik GYO TY KY |
%74 %16 %58 |
%87 %29 %58 |
0,0003 0,0005 0,9449 |
Yok Yok Yok |
K�saltmalar: GYO: genel yan�t oran�; TY: tam yan�t; KY: k�smi yan�t
�al��man�n idame faz�na randomize edilen hastalar i�in medyan g�zlem s�resi, idame randomizasyonundan itibaren 28 ayd�r. Rituximab ile idame tedavisi, sadece g�zlem koluna k�yasla, primer sonlanma noktas� olan PFS'de (idame randomizasyonundan n�kse, hastal�k ilerlemesine ya da �l�me kadar olan s�re) klinik olarak anlaml� ve istatistiksel olarak belirgin d�zelme ile sonu�lanm��t�r (p<0,000l, log-s�ra testi). Medyan PFS, rituximab idame kolunda 42,2 ayken g�zlem kolunda 14,3 ayd�r. Cox regresyon analizi kullan�ld���nda, hastal�k ilerlemesi ya da �l�m riski, rituximab idame tedavisi ile g�zleme g�re %61 oran�nda azalm��t�r (GA; %45-%72). 12 ayda Kaplan-Meier y�ntemiyle hesaplanan progresyonsuz oranlar, rituximab idame grubunda %78 iken g�zlem grubunda %57'dir. Genel sa�kal�m analizi, rituximab idamesinin, g�zleme g�re belirgin fayda sa�lad���n� kan�tlam��t�r (p=0,0039 log-s�ra testi). Rituximab idame tedavisi, �l�m riskini %56 azaltm��t�r (GA; %22-%75).
Tablo 9: �dame faz�: Rituximab ile g�zlem gruplar�n�n kar��la�t�rmal� etkililik sonu�lar�na genel bak�� (28 ayl�k medyan g�zlem s�resi)
Etkililik Parametresi | Olaya Kadar Medyan S�renin (ay) Kaplan-Meier Y�ntemiyle Hesaplanmas� |
Risk Azalt�m� | ||
G�zlem (N = 167) | Rituximab (N=167) | Log-s�ra p de�eri | ||
Progresyonsuz sa�kal�m (PFS) | 14,3 | 42,2 | <0,0001 | %61 |
Genel sa�kal�m (OS) | NR | NR | 0,0039 | %56 |
Yeni lenfoma tedavisine kadar ge�en s�re | 20,1 | 38,8 | <0,0001 | %50 |
Hastal�ks�z sa�kal�m | 16,5 | 53,7 | 0,0003 | %67 |
Alt Grup Analizi |
|
|
|
|
PFS |
|
|
|
|
CHOP | 11,6 | 37,5 | <0,0001 | %71 |
R-CHOP | 22,1 | 51,9 | 0,0071 | %46 |
TY | 14,3 | 52,8 | 0,0008 | %64 |
KY | 14,3 | 37,8 | <0,0001 | %54 |
OS |
|
|
|
|
CHOP | NR | NR | 0,0348 | %55 |
R-CHOP | NR | NR | 0,0482 | %56 |
NR: ula��lamam��t�r; : sadece TY'ye ula�an hastalar i�in
Rituximab idame tedavisinin faydas�, ind�ksiyon rejimi (CHOP ya da R-CHOP) ya da ind�ksiyon tedavisine verilen yan�tlar�n niteli�iyle (TY ya da KY) ilgili olmaks�z�n t�m alt gruplarda analiz edilmi�tir (Tablo 9). Rituximab idame tedavisi, CHOP ind�ksiyon tedavisine yan�t veren hastalarda (medyan PFS 37,5 aya kar��l�k 11,6 ay, p<0,0001) oldu�u kadar R-CHOP ind�ksiyon tedavisine yan�t veren hastalarda da (medyan PFS 51,9 aya kar��l�k 22,1 ay, p=0,0071) medyan PFS'yi �nemli �l��de uzatm��t�r. Alt gruplar k���k olsa da, rituximab idame tedavisi genel sa�kal�m a��s�ndan hem CHOP'a yan�t veren hastalarda hem de R-CHOP'a yan�t veren hastalarda klinik a��dan anlaml� fayda sa�lam��t�r, bu g�zlemi do�rulamak i�in daha uzun s�reli takip gereklidir.
Yeti�kinlerde diff�z b�y�k B h�creli Hodgkin-d��� lenfoma (DBBHL)
Randomize, a��k etiketli bir �al��mada, diff�z b�y�k B h�creli lenfomas� olan �nceden tedavi g�rmemi�, ya�lar� 60 ile 80 aras� de�i�en 399 ya�l� hastaya, sekiz siklus boyunca her �� haftada bir standart CHOP kemoterapisi (1. G�nde siklofosfamid 750 mg/m, doksorubisin 50 mg/m,
1. G�n maksimum 2 mg'a kadar, vinkristin 1,4 mg/m ve 1-5. G�nlerde prednizolon 40 mg/m/g�n) veya 375 mg/m rituximab + CHOP (R-CHOP) verilmi�tir. Rituximab tedavi siklusunun birinci g�n�nde uygulanm��t�r.
Nihai etkililik analizi randomize edilen t�m hastalar� (197 CHOP, 202 R-CHOP) kapsam��t�r ve ortalama izleme s�resi yakla��k 31 ayd�r. �ki tedavi grubu, ba�lang�� d�zeyi �zellikleri ve hastal�k durumu bak�m�ndan iyi dengelenmi�tir. Nihai analiz, R-CHOP tedavisinin olays�z ge�en sa�kal�m s�resini (primer etkililik parametresi, buradaki olaylar �l�m, n�ks veya lenfoma
ilerlemesi ya da yeni bir anti-lenfoma tedavisinin tesis edilmesidir) �nemli oranda uzatt���n� do�rulam��t�r (p=0,0001). Medyan olays�z sa�kal�m s�resine ili�kin Kaplan-Meier tahminlerine g�re, CHOP kolundaki 13 ay ile, R-CHOP kolunda 35 ay�n kar��la�t�r�lmas� riskin %41 azald���n� g�stermektedir. 24. Ayda, genel sa�kal�ma ili�kin tahminler CHOP kolundaki
%57,4'l�k orana k�yasla R-CHOP kolunda %68,2 olarak bulunmu�tur. Medyan 60 ayl�k izleme s�resi ile ger�ekle�tirilen daha sonraki bir genel sa�kal�m s�resi analizi, R-CHOP tedavisinin CHOP tedavisinden daha yararl� oldu�unu do�rulam�� (p=0,0071) ve riskin %32 azald���n� g�stermi�tir.
T�m sekonder parametrelerin analizi (yan�t oranlar�, progresyonsuz sa�kal�m, hastal�ks�z sa�kal�m, yan�t s�resi), CHOP ile kar��la�t�r�ld���nda R-CHOP tedavisinin etkisini do�rulam��t�r. 8. Siklustan sonra tam yan�t oran�, R-CHOP grubunda %76,2 ve CHOP grubunda
%62,4 bulunmu�tur (p=0,0028). Hastal���n ilerleme riski %46 ve n�ks riski %51 oran�nda azalt�lm��t�r.
T�m hasta alt gruplar�nda (cinsiyet, ya�, ya�a g�re ayarlanm�� IPI, Ann Arbor evresi, ECOG, β2 Mikroglobulin, LDH, alb�min, B semptomlar�, b�y�k hacimli hastal�k, ekstranodal b�lgeler, kemik ili�i tutulumu), olays�z sa�kal�m ve genel sa�kal�ma ili�kin risk oranlar� (R-CHOP'a kar��l�k CHOP) s�ras�yla 0,83 ve 0,95'den daha az bulunmu�tur. Ya�a g�re ayarlanm�� IPI'ye g�re R-CHOP hem y�ksek hem de d���k risk ta��yan hastalarda, sonu�ta ula��lan iyile�me d�zeyiyle ili�kili bulunmu�tur.
Klinik laboratuvar bulgular�
�nsan anti-fare antikoru (HAMA) a��s�ndan de�erlendirilen 67 hastan�n hi�biri i�in yan�t bildirilmemi�tir. �laca kar�� antikor (ADA) a��s�ndan de�erlendirilen 356 hastan�n %1,1'i (4 hasta) pozitif ��km��t�r.
Kronik lenfositik l�semi
A��k etiketli randomize iki �al��mada, daha �nce tedavi g�rmemi� toplam 817 KLL hastas� ve 552 relaps/refrakter KLL hastas�, 6 siklus i�in 4 haftada bir FC kemoterapi (fludarabin 25 mg/m, siklofosfamid 250 mg/m, 1-3. G�nler) veya FC ile kombinasyon halinde rituximab (R- FC) alacak �ekilde randomize edilmi�tir. Rituximab, ilk siklus s�ras�nda kemoterapiden bir g�n �nce 375 mg/m dozunda ve sonraki her tedavi siklusunun 1. G�n�nde 500 mg/m dozunda uygulanm��t�r. Relaps/refrakter KLL'de �nceden monoklonal antikorlar ile tedavi edilmi� veya fludarabin ya da herhangi bir n�kleozid analo�una refrakter olan hastalar (en az 6 ay i�in k�smi remisyon g�sterememe ba�ar�s�zl��� olarak tan�mlanm��t�r) �al��maya dahil edilmemi�tir. Etkililik i�in birinci basamak �al��mas�nda (Tablo 10a) ve (Tablo 10b) toplam 810 hasta (403 R-FC, 407 FC), relaps/refrakter �al��mas�nda da (Tablo 11) 552 hasta (276 R-FC, 276 FC) analiz edilmi�tir.
Birinci basamak �al��mas�nda 48,1 ayl�k medyan g�zlem s�resinden sonra medyan PFS, R- FC grubunda 55 ay ve FC grubunda 33 ay olmu�tur (p<0,0001, log-s�ra testi). Genel sa�kal�m analizi, yaln�zca FC kemoterapisi kullan�lan kola g�re, R-FC kolu i�in anlaml� bir fayda g�stermi�tir (p=0,0319, log-s�ra testi) (Tablo 10a). PFS a��s�ndan fayda, ba�lang��taki hastal�k riskine g�re (yani Binet A-C evreleri) (Tablo 10b) analiz edilen hasta alt gruplar�n�n �o�unda tutarl� olarak g�zlenmi�tir.
Tablo 10a: Kronik lenfositik l�seminin birinci basamak tedavisi
Tek ba��na FC'ye k�yasla rituximab+FC i�in etkililik sonu�lar�na genel bak�� – 48,1 ayl�k medyan g�zlem s�resi
Etkililik parametresi | Olaya kadar ge�en medyan s�re i�in Kaplan-Meier tahmini (Ay) | Risk azalt�m� | ||
FC (N = 409) | R-FC (N=408) | Log-s�ra p de�eri | ||
Progresyonsuz sa�kal�m (PFS) | 32,8 | 55,3 | <0,0001 | %45 |
Genel sa�kal�m | NR | NR | 0,0319 | %27 |
Olays�z sa�kal�m | 31,3 | 51,8 | <0,0001 | %44 |
Yan�t oran� (TY, nKY veya KY) TY oranlar� | %72,6
%16,9 | %85,8
%36 | <0,0001
<0,0001 | n.a.
n.a. |
Yan�t s�resi* | 36,2 | 57,3 | <0,0001 | %44 |
Hastal�ks�z sa�kal�m (DFS)** |
48,9 |
60,3 |
0,0520 |
%31 |
Yeni tedaviye kadar ge�en s�re | 47,2 | 69,7 | <0,0001 | %42 |
Yan�t oran� ve TY oranlar� Ki-kare Testi kullan�larak analiz edilmi�tir. NR: ula��lmad�; n.a.: ge�erli de�ildir.
*: Yaln�zca TY, nKY veya KY elde edilen hastalar i�in ge�erlidir.
**: Yaln�zca TY elde edilen hastalar i�in ge�erlidir.
Tablo 10b: Kronik lenfositik l�seminin birinci basamak tedavisi
Binet evresine g�re (ITT) progresyonsuz sa�kal�m tehlike oran� - medyan g�zlem s�resi 48,1 ay
Progresyonsuz sa�kal�m (PFS) | Hasta say�s� | Tehlike Oran� (%95 GA) | p-de�eri (Wald testi, ayarlanmam��) | |
FC | R-FC | |||
Binet evre A | 22 | 18 | 0,39 (0,15; 0,98) | 0,0442 |
Binet evre B | 259 | 263 | 0,52 (0,41; 0,66) | <0,0001 |
Binet evre C | 126 | 126 | 0,68 (0,49; 0,95) | 0,0224 |
GA: G�ven aral���
Relaps/refrakter �al��mas�nda, R-FC grubunda medyan progresyonsuz sa�kal�m (primer sonlan�m noktas�) 30,6 ay iken FC grubunda 20,6 ayd�r (p=0,0002, log-s�ra testi). PFS a��s�ndan fayda, ba�lang��taki hastal�k riskine g�re analiz edilen hasta alt gruplar�n�n neredeyse hepsinde g�zlenmi�tir. R-FC kolunda FC koluna k�yasla, genel sa�kal�mda (OS) az fakat anlaml� olmayan bir art�� bildirilmi�tir.
Tablo 11: Relaps/refrakter kronik lenfositik l�seminin tedavisi - Tek ba��na FC'ye k�yasla rituximab-FC i�in etkililik sonu�lar�na genel bak�� (medyan g�zlem s�resi 25,3 ay)
Etkililik Parametresi | Olaya Kadar Ge�en Medyan S�re i�in Kaplan-Meier Tahmini (Ay) | Risk azalt�m� | ||
FC (N = 276) | R-FC (N=276) | Log-s�ra p de�eri | ||
Progresyonsuz sa�kal�m (PFS) | 20,6 | 30,6 | 0,0002 | %35 |
Genel sa�kal�m | 51,9 | ula��lamad� | 0,2874 | %17 |
Olays�z sa�kal�m | 19,3 | 28,7 | 0,0002 | %36 |
Yan�t oran� (TY, nKY veya KY) | %58,0 | %69,9 | 0,0034 | uygulanabilir de�il |
TY oranlar� | %13,0 | %24,3 | 0,0007 | uygulanabilir de�il |
Yan�t s�resi* | 27,6 | 39,6 | 0,0252 | %31 |
Hastal�ks�z sa�kal�m (DFS)** | 42,2 | 39,6 | 0,8842 | %-6 |
Yeni KLL tedavisine kadar ge�en s�re | 34,2 | ula��lamad� | 0,0024 | %35 |
Yan�t oran� ve TY oranlar� Ki-kare Testi kullan�larak analiz edilmi�tir.
*: Yaln�zca TY, nKY, KY elde edilen hastalar i�in ge�erlidir.
**: Yaln�zca TY elde edilen hastalar i�in ge�erlidir.
�nceden tedavi edilmemi� ve/veya relaps/refrakter KLL hastalar�n�n tedavisinde di�er kemoterapi rejimleriyle (CHOP, FCM, PC, PCM, bendamustin ve kladribin dahil) kombinasyon halinde rituximab kullan�lan di�er destekleyici �al��malardan elde edilen sonu�lar, hafif y�kselmi� toksisiteye (�zellikle miyelotoksisite) ra�men, PFS oranlar� a��s�ndan faydal� y�ksek genel yan�t oranlar� ortaya koymu�tur. Bu �al��malar rituximab�n herhangi bir kemoterapi ile kullan�m�n� desteklemektedir.
Daha �nce rituximab ile tedavi edilmi� yakla��k 180 hastaya ait veriler klinik fayday� ortaya koymu�tur (TY dahil) ve bu veriler rituximab ile yeniden tedaviyi destekler niteliktedir.
Pediyatrik pop�lasyon
Daha �nce tedavi edilmemi� ileri evre CD20 pozitif DLBCL/BL/BAL/BLL'si olan pediyatrik hastalarda rituximab ile beraber veya rutiximab olmaks�z�n Lenfoma Malign B (LMB) kemoterapi (kortikosteroidler, vinkristin, siklofosfamid, y�ksek-doz metotreksat, sitarabin, doksorubisin, etoposid ve ��l� ila� [metotreksat/sitarabin/kortikosteroid] intratekal terapi) i�in �ok merkezli, a��k-etiketli, randomize bir �al��ma yap�lm��t�r. �leri evre, y�kselmi� LDH seviyesi (“B-y�ksek”) [LDH > yeti�kin normal de�erlerinin geleneksel �st s�n�r�n�n iki kat� (> Nx2)] ile beraber Evre III ya da herhangi bir Evre IV veya BAL olarak tan�mlanm��t�r. Hastalar LMB kemoterapi veya LMB �emas�na g�re LMB kemoterapisi ile kombine alt� i.v. rituximab inf�zyonunu (iki ind�ksiyon k�r�n�n her birinde iki tane ve iki konsolidasyon k�r�n�n her
birinde bir tane) 375 mg/m BSA dozunda almak �zere randomize edilmi�tir. Toplamda randomize edilmi� 328 hasta etkililik analizlerine dahil edilmi�tir, bu hastalar i�inde LMB kemoterapi ile kombine rituximab alan bir hasta 3 ya��n alt�ndayd�.
�ki tedavi kolu, LMB (LMB kemoterapi) ve R-LMB (rituximab ile LMB kemoterapi), ba�lang�� karakteristikleri a��s�ndan iyi dengelenmi�ti. Hastalar�n medyan ya�� LMB kolunda ve R-LMB kolunda s�ras�yla 7 ve 8 y�ld�. Hastalar�n yakla��k yar�s� (LMB kolunda %50,6 ve R-LMB kolunda %49,4) Grup B'deydi, her iki kolda %39,6's� Grup C1'deydi ve LMB ve R-LMB kollar� i�in s�ras�yla %9,8'i ve %11'i Grup C3'deydi. Murphy evrelemesine g�re, hastalar�n �o�u ya BL evre III (LMB kolunda %45,7 ve R-LMB kolunda %43,3) ya da BAL, SSS negatif (LMB kolunda %21,3 ve R-LMB kolunda %24,4)'ti. Hastalar�n yar�s�ndan az�nda (her iki kolda da %45,1) kemik ili�i tutulumu vard� ve hastalar�n �o�unda (LMB kolunda %72,6 ve R-LMB kolunda %73,2) SSS tutulumu yoktu. Primer etkililik sonlan�m noktas� Olays�z Sa�kal�m'd� (EFS), burada olay, hangisi �nce meydana gelirse gelsin, hastal�k ilerlemesi, relaps, ikinci malignite, herhangi bir sebepten �l�m veya ikinci CYVE k�r�nden sonra canl� h�cre kald���n�n saptanmas� ile do�rulanan tedaviye yan�t-olmamas� durumlar�n�n meydana gelmesi olarak tan�mlanm��t�. Sekonder etkililik sonlan�m noktalar� ise Genel Sa�kal�m ve Tam Remisyon olmu�tur.
Yakla��k 1 y�ll�k medyan takip s�resinde, �nceden tan�mlanm�� ara analizlerde, EFS primer sonlan�m noktas�nda klinik olarak alakal� d�zelmeler g�r�lm��t�r; 1-y�ll�k oran tahminleri R- LMB kolunda %94,2 (%95 GA, %88,5 - %97,2) olurken LMB kolunda %81,5 (%95 GA, %73,0
- %87,8) ve ayarlanm�� Cox Risk Regresyonu (HR) 0,33 (%95GA, 0,14 – 0,79) olmu�tur. Ba��ms�z Veri �zleme Komisyonu'nun bu sonuca dayanan tavsiyesi �zerine, randomizasyon durdurulmu� ve LMB kolundaki hastalar�n rituximab almak �zere ge�i� yapmalar�na izin verilmi�tir.
Randomize edilmi� 328 hastada primer etkililik analizleri yap�lm��t�r, medyan takip s�resi 3,1 y�ld�r. Sonu�lar Tablo 12'de a��klanmaktad�r.
Tablo 12: Primer Etkililik Sonu�lar�na Genel Bak�� (ITT Pop�lasyonu)
Analiz | LMB (N = 164) | R-LMB (N = 164) |
Olays�z Sa�kal�m (EFS) | 28 olay | 10 olay |
Tek-y�nl� log-s�ra test p-de�eri 0,0006 | ||
Ayarlanm�� Cox HR 0,32 (%90 GA: 0,17; 0,58) | ||
3-y�l EFS oranlar� | %82,3 (%95 GA: %75,7; %87,5) | %93,9 (%95 GA: %89,1; %96,7) |
Genel Sa�kal�m (OS) | 20 �l�m | 8 �l�m |
Tek-y�nl� log-s�ra test p-de�eri 0,0061 | ||
Ayarlanm�� Cox HR 0,36 (%95 GA: 0,16; 0,81) | ||
3-y�l OS oranlar� | %87,3 (%95 GA: %81,2; %91,6) | %95,1 (%95 GA: %90,5; %97,5) |
Tam Remisyon | %93,6 (%95 GA: %88,2; %97,0) | %94,0 (%95 GA: %88,8; %97,2) |
Primer etkililik analizi, rituximab ilave edilmi� LMB kemoterapinin tek ba��na LMB kemoterapiye g�re EFS avantaj�n� g�stermi�tir; ulusal grup, histoloji ve terap�tik grup i�in ayarlanm�� Cox regresyon analizinden elde edilen EFS HR 0,32 (%90 GA: 0,17 – 0,58) olmu�tur. �ki tedavi grubunda Tam Remisyon elde eden hastalar�n say�s�nda b�y�k bir fark olmasada, LMB kemoterapiye rituximab ilavesinin sekonder sonlan�m noktas� olan Genel Sa�kal�m �zerine faydas� da g�sterilmi�tir; OS HR 0,36 (%95 GA: 0,16 – 0,81) olmu�tur.
Pediyatrik kullan�m bilgisi i�in Bkz. B�l�m 4.2.
Gran�lamat�z polianjiitis (Wegener's) (GPA) ve mikroskobik polianjiitis (MPA)'de klinik deneyim
Yeti�kin remisyon ind�ksiyonu
GPA/MPA klinik �al��mas�na (�al��ma 1), ciddi aktif GPA (%75) ve MPA's� (%24) olan 15 ya� ve �zeri toplam 197 hasta dahil edilerek aktif kar��la�t�rmal�, randomize, �ift-k�r, �ok merkezli, non-inferior bir �al��mada tedavi edilmi�tir.
Hastalar 3-6 ay boyunca her g�n oral siklofosfamid (2 mg/kg/g�n) veya 4 hafta boyunca haftada bir rituximab (375 mg/m) almak �zere 1:1 oran�nda randomize edilmi�lerdi. Siklofosfamid kolundaki t�m hastalar takip s�resince azatioprin idame tedavisi alm��t�. Her iki koldaki hastalar 1 ila 3 g�n boyunca g�nde 1.000 mg pulse intraven�z (i.v.) metilprednizolon (veya e�de�er-dozda ba�ka bir glukokortikoid) ve ard�ndan oral prednizon (l mg/kg/g�n, en fazla 80 mg/g�n) alm��t�. Prednizon azalt�m� �al��ma tedavisinin ba�lamas�ndan itibaren 6 ayda tamamlanmal�yd�.
Primer sonu� �l��t� 6. Ayda tam remisyon sa�lanmas�yd� ve bu da Wegener Gran�lomatozisi i�in Birmingham Vask�lit Aktivite Skoru'nun (BVAS/WG) “0” olmas� ve glukokortikoid kullan�m�n�n b�rak�lmas� olarak tan�mlanmaktayd�. Tedavi fark� i�in �nceden belirlenen non- inferiorite marjini % 20'ydi. Bu �al��ma, 6. Ayda tam remisyon (CR) a��s�ndan siklofosfamide kar�� rituximab�n non-interferiotesini g�stermi�ti (Tablo 13).
Etkililik hem GPA ve MPA te�hisi yeni konan hastalarda hem de n�kseden hastal��� olan hastalarda g�zlenmi�tir (Tablo 14).
Tablo 13: 6. Ayda tam remisyona eri�en hastalar�n y�zdesi (tedavi meyilli pop�lasyon*)
| Rituximab (n = 99) | Siklofosfamid (n = 98) | Tedavi fark� (Rituximab – Siklofosfamid) |
Oran |
%63,6 |
%53,1 | %10,6 %95,1 GA (%-3,2, % 24,3) |
GA = g�ven aral���.
* En k�t� durum modeli
| Rituximab | Siklofosfamid | Fark (%95 GA) |
T�m hastalar | n=99 | n=98 |
|
Yeni te�his koyulmu� hastalar |
n=48 |
n=48 |
|
Relaps g�r�len hastalar |
n=51 |
n=50 |
|
Tam remisyon | |||
T�m hastalar | %63,6 | %53,1 | %10,6 (-3,2, 24,3) |
Yeni te�his | %60,4 | %64,6 | %-4,2 |
koyulmu� |
|
| (-23,6, 15,3) |
Relaps g�r�len | %66,7 | %42,0 | %24,7 |
hastalar |
|
| (5,8, 43,6) |
Tablo 14: Hastal�k durumuna g�re 6 aydaki tam remisyon
![]()
Verileri eksik olan hastalar i�in en k�t� durum modeli ge�erlidir. 12 ve 18. Aylarda tam remisyon (CR)
Rituximab grubunda hastalar�n %48'i 12 ayda ve %39'u 18 ayda CR'ye ula�m��t�r. Siklofosfamid (ve ard�ndan tam remisyonun idamesi i�in azatioprin) ile tedavi edilen hastalarda, hastalar�n %39'u 12 ayda ve %33'� 18 ayda CR'ye ula�m��t�r. 12. Aydan 18. Aya kadar rituximab grubunda 8 relaps g�r�l�rken siklofosfamid grubunda d�rt relaps g�r�lm��t�r.
Laboratuvar de�erlendirmeleri
Remisyon tedavisinin ind�ksiyonu �al��mas�nda rituximab ile tedavi edilen toplam 99 hastan�n 23'� (%23) 18 aya kadar ADA a��s�ndan pozitif bulunmu�tur. Rituximab ile tedavi edilen 99 hastan�n hi�biri �al��ma ba�lang�c�ndaki taramada ADA a��s�ndan pozitif de�ildir. Remisyon tedavisinin ind�ksiyonu �al��mas�nda ADA varl���n�n g�venlilik veya etkililik �zerinde belirgin bir e�ilimi veya olumsuz bir etkisi olmam��t�r.
Pemfigus vulgaris'te klinik deneyim PV �al��mas� 1 (�al��ma ML22196)
Rituximab�n k�sa-s�reli, d���k-dozlu glukokortikoid (prednizon) tedavisi ile kombine kullan�m�n�n etkililik ve g�venlili�i, yeni tan� konmu�, orta ila �iddetli pemfigusu (74 pemfigus vulgaris [PV] ve 16 pemfigus foliaseus [PF]) olan hastalarda yap�lan bu randomize, a��k- etiketli, kontroll�, �ok-merkezli �al��mada de�erlendirilmi�tir. Hastalar 19 ve 79 ya� aral���ndayd� ve daha �nce pemfigus i�in tedavi g�rmemi�lerdi. PV pop�lasyonunda, Harman kriterleri taraf�ndan tan�mlanm�� hastal�k �iddetine g�re rituximab grubunda 5 hastan�n (%13) ve standart prednizon grubunda 3 hastan�n (%8) orta �iddette hastal��� ve 33 hastan�n (%87) ve standart prednizon grubunda 33 hastan�n (%92) �iddetli hastal��� vard�.
Hastalar ba�lang��ta hastal�k �iddetine (orta ila �iddetli) grupland�r�lm��lard� ve rituximab ve d���k-doz prednizon ya da standart doz prednizon alacak �ekilde 1:1 oran�nda randomize edilmi�lerdi. Rituximab grubuna randomize edilen hastalar �al��man�n 1. G�n� 1.000 mg'l�k ilk rituximab intraven�z inf�zyonu ile birlikte orta �iddette hastal��� olanlar 3 ay i�erisinde azalt�larak kesilecek olan 0,5 mg/kg/g�n oral prednizon, ya da hastal��� �iddetli olanlar 6 ay i�erisinde azalt�larak kesilecek olan 1 mg/kg/g�n oral prednizon alm��lard�r. �al��man�n 15.
G�n� ikinci bir 1.000 mg'l�k intraven�z inf�zyon uygulanm��t�r. Rituximab�n idame dozlar� 12. ve 18. Aylarda 500 mg dozunda uygulanm��t�r. Standart-doz prednizon grubuna randomize edilen orta �iddette hastal��� olan hastalar ba�lang��ta 1 mg/kg/g�n oral prednizonu 12 ay i�erisinde azalt�larak kesilmek �zere ya da �iddetli hastal��� olan hastalar 1,5 mg/kg/g�n oral prednizonu 18 ay i�erisinde azalt�larak kesilmek �zere alm��lard�r. Rituximab grubunda relaps ya�ayan hastalar ilave bir 1.000 mg rituximab inf�zyonu ile beraber yeniden prednizolon almaya ba�layabilmi�lerdir ya da birlikte kullan�lan prednizon dozu artt�r�labilmi�tir. �dame ya da relaps inf�zyonlar� bir �nceki inf�zyonun ard�ndan en az 16 hafta sonra uygulanm��t�r.
�al��man�n primer objektifi en az iki ay s�resince prednizon tedavisi kullanmadan (CRoff ≥2 ay) 24. Ayda tam remisyondu (tam epitelizasyon ve yeni ve/veya �nceden mevcut lezyonlar�n olmamas�).
PV �al��ma 1 sonu�lar�
�al��ma rituximab ve d���k-doz prednizonun standart doz prednizona g�re, PV hastalar�nda
24. Ayda CRoff ≥2 ay hedefinin ba�ar�lmas�nda istatistiksel olarak anlaml� sonu�lar g�stermi�tir (Bkz. Tablo 14).
Tablo 14 24. Ayda kortikoid tedavisi olmadan en az 2 ay s�resince tam remisyonu ba�arm�� PV hastalar�n�n y�zdesi (Tedavi Meyilli Pop�lasyon – PV)
| Rituximab + Prednizon N = 38 | Prednizon N = 36 | p-de�eri | %95 GA |
Yan�t verenlerin say�s� | 34 (%89,5) | 10 (%27,8) | <0,0001 | %61,7 (38,4, 76,5) |
(yan�t oran� [%]) |
|
|
|
|
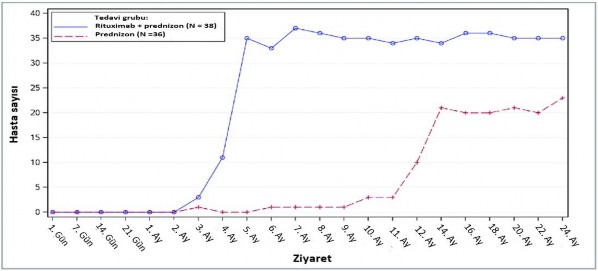
24 ay s�ren tedavi periyodu boyunca, rituximab art� d���k-doz prednizon hastalar�nda prednizon tedavisini b�rakm�� olan ya da minimal tedavi alan (g�nl�k prednizon dozu 10 mg veya daha az) hastalar�n say�s�n�n standart doz prednizon hastalar�n�n say�s� ile k�yaslanmas� rituximab�n steroid-azalt�m� etkisini g�stermi�tir (�ekil 1).
�ekil 1: Zaman i�erisinde kortikosteroidi b�rakan ya da minimal kullanan (≤10 mg/g�n) hastalar�n say�s�
Post-hoc geriye d�n�k laboratuvar de�erlendirmesi
Rituximab ile tedavi edilen PV hastalar�n�n toplam 19/37'si (%56) 18. aya kadar ADA antikorlar� a��s�ndan pozitif sonu� vermi�lerdir. Rituximab ile tedavi edilen PV hastalar�nda ADA olu�umunun klinik ili�kisi net de�ildir.
PV �al��mas� 2 (�al��ma WA29330)
Randomize, �ift-k�r, �ift-sa��r maskelenmi�, aktif- kontroll�, �ok-merkezli bir �al��mada, orta ila �iddetli PV hastal��� olan ve �al��maya kat�l�rken 60-120 mg/g�n dozunda oral prednizon veya e�de�erini (1,0 – 1,5 mg/kg/g�n) kullanan ve zaman i�erisinde azalt�larak 1. G�nde 60 veya 80 mg/g�n dozuna ula�an hastalarda, rituximab�n etkililik ve g�venlili�i mikofenolat mofetil (MMF) ile k�yaslanm��t�r. Hastalar�n ge�mi� 24 ay i�erisinde PV te�hisi ald��� ve hastal�klar�n�n orta ila �iddetli oldu�una dair kan�tlar (Pemfigus Hastal��� Alan Indeksi, PDAI, aktivite skoru ≥ 15) teyit edilmi�tir.
135 hasta 24. Haftada prednizon dozunun 0 mg/g�ne do�ru azalt�larak kesilmesi amac�yla, 60 veya 80 mg oral prednison ile kombine olarak 1. G�n, 15. G�n, 24. Hafta ve 26. Hafta'da uygulanacak 1.000 mg rituximab ile tedaviye ya da 52 hafta boyunca 2 g/g�n dozunda oral MMF ile tedaviye randomize edilmi�tir.
Bu �al��man�n primer etkililik amac� 52. Haftada 0 mg/g�n dozunda prednizon veya e�de�erini kullan�rken hi� yeni aktif lezyon olmadan lezyonlar�n iyile�mesinin sa�lanmas� (�rn. PDAI aktivite skoru 0) ve 52 haftal�k tedavi periyodunda, bu yan�t�n ardarda en az 16 hafta boyunca devam ettirilmesi olarak tarif edilen devaml� tam remisyonun elde edilmesinde rituximab�n etkilili�inin MMF ile kar��la�t�rmas�n� de�erlendirmekti.
PV �al��mas� 2 sonu�lar�
Bu �al��ma PV hastalar�nda 52. Haftada CRoff kortikosteroid ≥16 hafta hedefine ula��lmas�nda kademeli olarak azalt�larak b�rak�lan oral kortikosteroid k�r� ile beraber kullan�lan rituximab�n MMF'ye �st�nl���n� g�stermi�tir (Tablo 15). mITT pop�lasyonundaki hastalar�n �o�unlu�u yeni te�his konmu� hastalard� (%74) ve hastalar�n %26's�n�n hastal��� �nceden mevcuttu (hastal���n s�resi ≥6 ay ve PV i�in daha �nce tedavi alm��lard�).
Tablo 15 52. Haftada 16 Hafta veya Daha Uzun S�re Boyunca Kortikosteroid Tedavisi Almadan Devaml� Tam Remisyonda Kalan PV Hastalar�n�n Y�zdesi
| Rituximab (N=62) | MMF (N=63) | Fark (%95 GA) | p-de�eri |
Yan�t verenlerin say�s� (yan�t oran� [%]) Yeni te�his konmu� hastalar Hastal��� �nceden mevcut hastalar | 25 (%40,3) | 6 (%9,5) | %30,80 (%14,70, %45,15) | <0,0001 |
19 (%39,6) | 4 (%9,1) |
|
| |
6 (%42,9) | 2 (%10,5) |
|
| |
MMF = Mikofenolat mofetil GA = G�ven aral��� Yeni te�his konmu� hastalar = hastal�k s�resi <6 ay veya PV i�in daha �nce tedavi g�rmemi� olmak Hastal��� �nceden mevcut hastalar = hastal�k s�resi ≥6 ay ve PV i�in daha �nce tedavi g�rm�� olmak p-de�eri i�in Cochran-Mantel-Haenszel testi kullan�lm��t�r. | ||||
Sekonder parametrelerin (k�m�latif oral kortikosteroid dozu, hastal�k alevlenmelerinin toplam say�s� ve sa�l�kla alakal� olarak Dermatoloji Hayat Kalite �ndeksi ile �l��len hayat kalitesinde de�i�im dahil) analizi rituximab�n MMF'ye k�yasla istatistiksel olarak anlaml� sonu�lar ortaya koydu�unu do�rulam��t�r.
Glukokortikoide maruziyet
Rituximab ile tedavi edilen hastalarda k�m�latif oral kortikosteroid dozu anlaml� �l��de daha d���k olmu�tur. 52. Haftada medyan (min, maks) k�m�latif prednizon dozu, rituximab grubunda 2.775 mg (450, 22.180) olurken MMF grubunda 4.005 mg (900, 19.920) olmu�tur (p=0,0005).
Hastal�k alevlenmeleri
Rituximab ile tedavi edilen hastalarda hastal�k alevlenmelerinin toplam say�s� MMF ile tedavi edilen hastalardakine g�re anlaml� �l��de d���k olmu�tur (6'ya kar��l�k 44, p<0,0001) ve en az bir hastal�k alevlenmesi ya�ayan hasta say�s� daha az olmu�tur (%8,1'e kar��l�k %41,3).
Laboratuvar de�erlendirmeleri
52. Haftaya kadar, rituximab ile tedavi edilen PV hastalar�ndan toplamda 20/63'� (%31,7) (19'u tedavi sebebiyle ba�layan ve 1'i tedavi sebebiyle artan) ADA pozitif olarak test edilmi�tir. ADA varl���n�n PV �al��mas� 2'deki g�venlilik veya etkililik �zerine belirgin bir negatif etkisi olmam��t�r.
5.2. Farmakokinetik �zellikler
Genel �zelliklerEmilim
Hodgkin-d��� lenfoma
Tekli ajan olarak ya da CHOP tedavisi ile kombinasyon halinde tek veya �ok say�da rituximab inf�zyonu alan (uygulanan rituximab dozlar� 100 ile 500 mg/m aras�nda de�i�mi�tir) 298 Hodgkin-d��� lenfoma hastas�nda y�r�t�len bir pop�lasyon farmakokineti�i analizi temelinde, spesifik olmayan klerens (CL), B h�creleri veya t�m�r y�k�n�n katk�da bulunmas� olas� spesifik klerens (CL) ve santral kompart�man da��l�m hacmi (V) i�in tipik pop�lasyon tahminleri s�ras�yla 0,14 L/g�n, 0,59 L/g�n ve 2,7 L'dir. Rituximab�n hesaplanan medyan terminal eliminasyon yar�lanma �mr� 22 g�nd�r (6,1 ila 52 g�n aral���nda). 4 haftal�k doz boyunca intraven�z inf�zyon olarak 375 mg/m verilen 161 hastadan elde edilen verilerde ba�lang�� CD19-pozitif h�cre say�mlar� ve �l��lebilir t�m�r lezyonlar�n�n boyutu, rituximab�n CL'sindeki de�i�kenli�in bir k�sm�na katk�da bulunmu�tur. Daha y�ksek CD19-pozitif h�cre say�mlar�na veya t�m�r lezyonlar�na sahip hastalar daha y�ksek CL'ye sahiptir. Bununla birlikte, CD19-pozitif h�cre say�mlar� ve t�m�r lezyon boyutu i�in d�zeltme sonras� CLi�in bireyler aras� de�i�kenli�in b�y�k k�sm� devam etmi�tir. Vv�cut y�zey alan� (BSA) ve CHOP tedavisine g�re de�i�mi�tir. S�ras�yla BSA'daki aral�k (1,52 ila 2,32 m) ve e�zamanl� CHOP tedavisinin katk�da bulundu�u V'deki bu de�i�kenlik (%27,1 ve %19,0) nispeten k���kt�r. Ya�, cinsiyet ve D�nya Sa�l�k �rg�t� performans durumu rituximab�n farmakokineti�i �zerinde bir etkiye sahip de�ildir. Bu analiz test edilen kovaryatlardan herhangi biri ile rituximab dozunun ayarlanmas�n�n farmakokinetik de�i�kenli�inde anlaml� bir azalma ile sonu�lanmas�n�n beklenmedi�ini d���nd�rmektedir.
Daha �nce rituximab kullanmam�� Hodgkin-d��� lenfomal� 203 hastaya 4 doz boyunca haftada bir aral�klarla 375 mg/m dozda intraven�z inf�zyon olarak uygulanan rituximab, d�rd�nc� inf�zyonu takiben 486 mikrogram/mL'lik (77,5 ila 996,6 mikrogram/mL aral���nda) ortalama
bir Cvermi�tir. Rituximab son tedavinin tamamlanmas�ndan 3-6 ay sonra hastalar�n serumunda tespit edilebilmi�tir.
Rituximab�n Hodgkin-d��� lenfomal� 37 hastaya 8 doz boyunca haftada bir aralarla intraven�z inf�zyon olarak 375 mg/m'lik bir dozda uygulanmas�ndan sonra ortalama Cher bir ard���k inf�zyon ile artm�� olup, ilk inf�zyondan sonra ortalama 243 mikrogram/mL (16-582 mikrogram/mL aral���nda) ve sekizinci inf�zyondan sonra 550 mikrogram/mL (171-1.177 mikrogram/mL aral���nda) aras�nda de�i�mi�tir.
6 siklus CHOP kemoterapisi ile kombinasyon halinde 375 mg/m'lik 6 inf�zyon olarak uyguland���nda rituximab�n farmakokinetik profili, tek ba��na rituximab ile g�r�lene benzerdir.
Kronik lenfositik l�semi
Rituximab KLL hastalar�nda, fludarabin ve siklofosfamid ile kombinasyon halinde, i.v. inf�zyon olarak ilk siklusta 375 mg/m, sonraki 5 siklusun her birinde 500 mg/m'ye art�r�larak uygulanm��t�r. Be�inci 500 mg/m'lik inf�zyondan sonra ortalama C(n=15) 408 mikrogram/mL (97-764 mikrogram/mL aral���nda), ortalama terminal yar�lanma �mr� de 32 g�nd�r (14 ila 62 g�n aral���nda).
Gran�lomat�z polianjiitis (Wegener) (GPA) ve mikroskobik polianjiitis (MPA)
D�rt doz boyunca haftada bir kez 375 mg/m rituximab alm�� gran�lomat�z polianjiitis ve mikroskobik polianjiitis g�r�len 97 hastada pop�lasyon farmakokineti�i analizi temelinde hesaplanm�� ortalama terminal eliminasyon yar�lanma �mr� 23 g�nd�r (9 ila 49 g�n aral���nda). Rituximab ortalama klerensi ve da��l�m hacmi s�ras�yla 0,313 L/g�n (0,116 ila 0,726 L/g�n aral���nda) ve 4,50 L'dir (2,25 ila 7,39 L aral���nda). �lk 180 g�n boyunca maksimum konsantrasyon (C), 180. G�nde minimum konsantrasyon (C180) ve 180 g�n boyunca e�rinin alt�ndaki k�m�latif alan (EAA180) (medyan [aral�k]) s�ras�yla 372,6 (252,3-533,5) mikrogram/mL, 2,1 (0-29,3) mikrogram/mL ve 10.302 (3.653-21.874) mikrogram/mL/g�n'd�r. Bu hastalarda rituximab�n farmakokinetik parametreleri romatoid artrit hastalar�nda g�zlenene benzer g�r�nmektedir.
Pemfigus vulgaris
1. G�n, 15. G�n, 168. G�n ve 182. G�nde 1.000 mg dozda rituximab alan yeti�kin PV hastalar�ndaki farmakokinetik parametreler Tablo 16'da �zetlenmi�tir.
Tablo 16 PV �al��mas� 2'den Elde Edilen, Yeti�kin PV Hastalar�ndaki Pop�lasyon Farmakokineti�i
Parametre | �nf�zyon Siklusu | |
| 1.000 mg'l�k 1. Siklus 1. G�n ve 15. G�n N=67 | 1.000 mg'l�k 2. Siklus 168. G�n ve 182. G�n N=67 |
Terminal yar�lanma �mr� (g�n) Medyan (Aral�k) |
21,0 (9,3-36,2) |
26,5 (16,4-42,8) |
Klerens (L/g�n) Ortalama (Range) |
391 (159-1.510) |
247 (128-454) |
Da��l�m�n Santral Hacmi (L) |
|
|
Ortalama | 3,52 | 3,52 |
(Aral�k) | (2,48-5,22) | (2,48-5,22) |
�lk iki rituximab uygulamas�n� takiben (1. Siklusa denk gelen 1. G�nde ve 15. G�nde) PV'li hastalarda rituximab�n farmakokinetik parametrelerin GPA/MPA ve romatoid artritli hastalarda g�r�lenlere benzer olmu�tur. Son iki uygulaman�n ard�ndan (2. Siklusa denk gelen 168. G�nde ve 182. G�nde), rituximab klerensi azal�rken, da��l�m�n santral hacmi de�i�meden kalm��t�r.
Biyotransformasyon Hodgkin-d��� lenfoma Veri bulunmamaktad�r.
Kronik lenfositik l�semi Veri bulunmamaktad�r.
Gran�lomat�z polianjiitis (Wegener) (GPA) ve mikroskobik polianjiitis (MPA)
Veri bulunmamaktad�r.
Pemfigus vulgaris
Veri bulunmamaktad�r.
Da��l�m
Hodgkin-d��� lenfoma
Tek ajan olarak ya da CHOP tedavisi ile kombinasyon halinde tek veya �ok say�da rituximab inf�zyonu alan (uygulanan rituximab dozlar� 100 ila 500 mg/m aras�nda de�i�mi�tir) 298 Hodgkin-d��� lenfoma hastas�nda y�r�t�len bir pop�lasyon farmakokine�i analizine dayanarak, spesifik olmayan klerens (CL), muhtemelen B h�creleri veya t�m�r y�k�n�n katk�da bulundu�u spesifik klerens (CL) ve santral kompartman da��l�m hacmi (V) i�in tipik pop�lasyon tahminleri s�ras�yla 0,14 L/g�n, 0,59 L/ g�n ve 2,7 L'dir.
Kronik lenfositik l�semi Veri bulunmamaktad�r.
Gran�lomat�z polianjiitis (Wegener) (GPA) ve mikroskobik polianjiitis (MPA)
Rituximab ortalama klerensi ve da��l�m hacmi s�ras�yla 0,313 L/g�n (aral�k, 0,116 ila 0,726 L/g�n) ve 4,50 L'dir (aral�k, 2,25 ila 7,39 L).
Pemfigus vulgaris
Da��l�m�n santral hacmi ortalama 3,52 L (2,48-5,22)'dir. �lk iki rituximab uygulamas�n� takiben (1. Siklusa denk gelen 1. G�nde ve 15. G�nde) ve son iki uygulaman�n ard�ndan (2. Siklusa denk gelen 168. G�nde ve 182. G�nde), rituximab da��l�m�n�n santral hacmi de�i�meden kalm��t�r.
Eliminasyon Hodgkin-d��� lenfoma
Rituximab�n tahmin edilen medyan terminal eliminasyon yar�lanma �mr� 22 g�nd�r (6,1 ila 52 g�n aral���nda). Rituximab�n, 4 haftal�k dozlar halinde 375 mg/m i.v. inf�zyon olarak verildi�i 161 hastadan elde edilen verilerde CL'sindeki de�i�kenli�in bir k�sm�na ba�lang�� CD19- pozitif h�cre say�s� ve �l��lebilir t�m�r lezyonlar�n�n boyutu katk�da bulunmu�tur. Daha y�ksek CD19-pozitif h�cre say�s� ya da t�m�r lezyonlar� olan hastalar�n daha y�ksek CL'si olmu�tur. Bununla birlikte, CD19-pozitif h�cre say�s� ve t�m�r lezyonu boyutu i�in d�zeltme yap�ld�ktan sonra, CLi�in bireyler aras� de�i�kenli�in b�y�k k�sm� aynen kalm��t�r. V1, v�cut y�zey alan� (BSA) ve CHOP tedavisi ile de�i�mi�tir. S�ras�yla, BSA'daki aral�k (1,53 ila 2,32 m) ve e�zamanl� CHOP tedavisinin katk�da bulundu�u V1'deki (%27,1 ve %19,0) bu de�i�kenlik, g�receli olarak k���kt�r.
Kronik lenfositik l�semi
KLL hastalar�nda, fludarabin ve siklofosfamid ile kombinasyon halinde verilen be�inci 500 mg/m'lik i.v. inf�zyondan sonra ortalama terminal yar�lanma �mr� 32 g�nd�r (14 ila 62 g�n).
Gran�lamat�z polianjiitis (Wegener) (GPA) ve mikroskobik polianjiitis (MPA)
D�rt doz boyunca haftada bir kere 375 mg/m rituximab alan 97 GPA ve MPA hastas�ndaki verilerin pop�lasyon farmakokinetik analizine dayan�larak, tahmini medyan terminal eliminasyon yar� �mr� 23 g�nd�r (9 ila 49 g�n aral���nda). Rituximab ortalama klerensi 0,313 L/g�n (0,116 ila 0,726 L/g�n aral���nda) olarak bulunmu�tur.
Pemfigus vulgaris
�lk iki rituximab uygulamas�n� takiben (1. Siklusa denk gelen 1. G�nde ve 15. G�nde) medyan terminal yar�lanma �mr� 21.0 (9,3-36,2) g�nd�r. Son iki uygulaman�n ard�ndan (2. Siklusa denk gelen 168. G�nde ve 182. G�nde) medyan terminal yar�lanma �mr� 26,5 (16,4-42,8)
g�nd�r. 1. Siklusun ard�ndan ortalama klerens 391 L/g�n (159-1.510) olurken, 2. Siklus ard�ndan ortalama klerens azalarak 247 L/g�n (128-454) olmu�tur.
Do�rusall�k/do�rusal olmayan durum
D�rt �al��mada, iki hafta aral�klarla 1. g�n ve 15. g�nde uygulanan 500 mg ve 1.000 mg dozlardaki iki intraven�z rituximab inf�zyonunu takiben, rituximab farmakokineti�i ara�t�r�lm��t�r. Bu �al��malar�n hepsinde, rituximab farmakokineti�i ara�t�r�lan limitli doz aral��� i�in doz ile orant�l� olmu�tur.
Pediyatrik pop�lasyon DLBCL/BL/BAL/BLL
Emilim
Pediyatrik DLBCL/BL/BAL/BLL'nin �al���ld��� klinik �al��mada, farmakokinetik, 3 ya��ndan b�y�k 35 hastal�k bir alt grupta �al���lm��t�r. �ki ya� grubu (≥ 3 ila < 12 ya� ve ≥ 12 ya� ila < 18 ya�) aras�nda PK kar��la�t�r�labilir olmu�tur. Her iki ind�ksiyon siklusunda (siklus 1 ve 2) 375 mg/m dozunda uygulanan iki rituximab inf�zyonunu takip eden konsolidayon sikluslar�n�n (siklus 3 ve 4) herbirinde 375 mg/m dozunda uygulanan bir rituximab inf�zyonu sonras�nda maksimum konsantrasyon d�rd�nc� inf�zyondan (siklus 2) sonra en y�ksek olmu�tur; geometrik ortalama 347 mikrogram/mL olmu�tur, bunu maksimum konsantrasyonun daha d���k geometrik ortalamalar� takip etmi�tir (siklus 4: 247 mikrogram/mL). Bu doz rejimi ile,
vadi d�zeyleri s�rd�r�lm��t�r (geometrik ortalama 41,8 mikrogram/mL (predoz Siklus 2; 1 siklusun arkas�ndan), 67,7 mikrogram/mL (predoz Siklus 3, 2 siklusun ard�ndan) ve 58,5 mikrogram/mL (predoz Siklus 4, 3 siklusun ard�ndan).
Rituximab�n DLBCL/BL/BAL/BLL'li pediyatrik hastalardaki PK karateristikleri yeti�kin NHL hastalar�nda g�zlenene benzerdir.
≥ 6 ay'dan < 3 ya��na kadar olan grup i�in PK verisi mevcut de�ildir ancak, pop�lasyon PK tahminleri ≥ 3 ya� grubu ile k�yasland���nda bu ya� grubunda kar��la�t�r�labilir sistemik maruziyeti (EAA, Cvadi) destekler (Tablo 17). Ba�lang��taki daha k���k t�m�r b�y�kl���, daha d���k zamana ba��ml� klerens sebebiyle daha y�ksek maruziyet ile ili�kilidir ancak farkl� t�m�r b�y�kl�klerinden etkilenen sistemik maruziyet, etkili ve g�venlilik profili a��s�ndan kabul edilebilir maruziyet aral��� i�erisinde kalm��t�r.
Tablo 17: Pediyatrik DLBCL/BL/BAL/BLL'de Rituximab Doz Rejiminin Ard�ndan Tahmini PK Parametreleri
Ya� Grubu | ≥ 6 ay ila < 3 ya� | ≥ 3 ya� ila < 12 ya� | ≥ 12 ya� ila < 18 ya� |
C(mikrogram/mL) | 47,5 (0,01 – 179) | 51,4 (0,00 – 182) | 44,1 (0,00 – 149) |
EAA (mikrogram*g�n/mL) | 13.501 (278 – 31.070) | 11.609 (135 – 31.157) | 11.467 (110 – 27.066) |
Sonu�lar medyan olarak sunulmu�tur (min – maks); Cpredoz Siklus 4't�r. At�l�m
3 ya��ndan b�y�k pediyatrik hastalarda ortalama eliminasyon yar�lanma �mr� 26 g�n olmu�tur. Hastalardaki karakteristik �zellikler
B�brek veya karaci�er yetmezli�i olan hastalara ait farmakokinetik veri bulunmamaktad�r.
Rituximab�n farmakokineti�i �zerine ya�, cinsiyet, �rk ve D�nya Sa�l�k �rg�t� performans stat�s�n�n herhangi bir etkisi olmam��t�r. Bu analiz, test edilen e�de�i�ken fakt�rlerin herhangi biriyle rituximab�n doz ayarlamas�n�n, farmakokinetik de�i�kenlikte anlaml� bir azalmayla sonu�lanmas�n�n beklenmedi�ini belirtmektedir.
5.3. Klinik �ncesi g�venlilik verileri
Rituximab�n B h�crelerindeki CD20 antijenine y�ksek d�zeyde spesifik oldu�u g�sterilmi�tir. Sinomolgus maymunlar�nda yap�lan toksisite �al��malar�, periferik kanda ve lenfoid dokuda B h�crelerinin beklenen farmakolojik t�ketiminden ba�ka bir etki g�stermemi�tir.
100 mg/kg'a kadarki dozlarda (gestasyonun 20-50. g�nleri aras�nda tedavi) sinomolgus maymunlar� �zerinde geli�imsel toksisite �al��malar� yap�lm�� ve fet�s i�in rituximabdan kaynaklanan herhangi bir toksisite kan�t�n�n olmad��� g�sterilmi�tir. Ancak fet�s�n lenfoid organlar�nda B h�crelerinin doza ba�l� farmakolojik t�ketimi g�zlenmi� olup bu do�umdan sonra da devam etmi� ve buna etkilenen yeni do�an hayvanlarda IgG d�zeylerinde bir azalma da e�lik etmi�tir. B h�cre say�s�, bu hayvanlarda do�umu takip eden 6 ay i�erisinde normale d�nm�� ve imm�nizasyon reaksiyonunu riske atmam��t�r.
Mutajeniteyi ara�t�ran standart testler bu molek�l i�in uygun olmad���ndan yap�lmam��t�r. Rituximab�n karsinojenik potansiyelini belirlemek i�in uzun d�nem hayvan �al��malar� yap�lmam��t�r.
Rituximab�n fertilite �zerine etkilerini tespit etmek i�in spesifik �al��malar yap�lmam��t�r. Sinomolgus maymunlar�ndaki genel toksisite �al��malar�nda, erkek ya da di�ilerin �reme organlar� �zerine zararl� hi�bir etki g�zlenmemi�tir.
6. FARMAS�T�K �ZELL�KLER
6.1. Yard�mc� maddelerin listesi
Tri-sodyum sitrat dihidrat
6.2. Ge�imsizlikler
Rituximab ile polivinilklor�r veya polietilen torbalar veya inf�zyon seti aras�nda ge�imsizlik g�zlenmemi�tir.
6.3. Raf �mr�
A��lmam�� flakonlar:
36 ay.
Seyreltilmi� �r�nler:
Mikrobiyolojik a��dan, seyreltilmi� �r�nlerin hemen kullan�lmas� gerekir. Bu �r�nler hemen kullan�lmazsa, seyreltme i�lemi kontroll� ve valide edilmi� aseptik ko�ullar alt�nda ger�ekle�tirilmedi�i takdirde kullan�m s�ras�ndaki saklama s�releri ve kullan�m �ncesindeki ko�ullar kullan�c�n�n sorumlulu�undad�r ve normal olarak 2°C-8°C'de 24 saati a�mamal�d�r.
Ayr�ca PE/PVC inf�zyon torbas�nda seyreltilen �r�n (%0,9 sodyum klor�r veya %5'lik dekstrozun sudaki ��zeltisi i�erisinde) fiziksel ve kimyasal olarak 24 saat s�resince 2°C-8°C'de ve sonra da 12 saat s�resince 30°C alt�ndaki oda s�cakl���nda stabildir.
6.4. Saklamaya y�nelik �zel tedbirler
Flakonlar� 2°-8°C'de (buzdolab�nda) saklay�n�z. Dondurulmamal�d�r. Flakonlar� do�rudan g�ne� �����ndan korumak i�in ambalaj�nda saklay�n�z.
�r�n�n seyretmeden sonraki saklama ko�ullar� i�in Bkz. B�l�m 6.3.
6.5. Ambalaj�n niteli�i ve i�eri�i
10 mL'de 100 mg rituximab (10 mg/mL) i�eren, klorobutil kau�uk t�pal� �effaf Tip 1 cam flakon.
2 flakon i�eren ambalajlarda.
6.6. Be�eri t�bbi �r�nden arta kalan maddelerin imhas� ve di�er �zel �nlemler
TRUXIMA inf�zyonlar�, acil uygulama i�in t�m res�sitasyon olanaklar�n�n bulundu�u merkezlerde ve deneyimli bir onkolog/hematolo�un g�zetimi alt�nda uygulanmal�d�r.
TRUXIMA steril, koruyucu maddeler i�ermeyen, tek dozluk flakonlarda sunulur.
TRUXIMA'y� haz�rlamak i�in steril i�ne ve ��r�nga kullan�n. Gerekli miktarda TRUXIMA'y� aseptik ko�ullarda �ekiniz ve i�inde steril, pirojen bulundurmayan, %0,9'luk sodyum klor�r�n veya %5'lik dekstrozun sudaki ��zeltisinden bulunan bir inf�zyon torbas�nda (PE/PVC torba), hesaplanm�� olan 1 - 4 mg/mL'lik rituximab konsantrasyonuna ula��ncaya dek seyreltiniz. ��zeltiyi kar��t�rmak i�in, torbay� k�p�k olu�umunu �nleyecek �ekilde nazik�e ters �eviriniz. Haz�rlanan ��zeltinin steril oldu�undan emin olunmal�d�r. Bu ila� herhangi bir antimikrobiyal koruyucu veya bakteriyostatik ajan i�ermedi�i i�in aseptik teknikler uygulanmal�d�r.
Parenteral ila�lar uygulanmadan �nce, partik�ll� maddeler ve renk de�i�ikli�ine dikkat edilmelidir.
Mikrobiyolojik a��dan, seyreltilmi� �r�nlerin hemen kullan�lmas� gerekir. Bu �r�nler hemen kullan�lmazsa, seyreltme i�lemi kontroll� ve valide edilmi� aseptik ko�ullar alt�nda ger�ekle�tirilmedi�i takdirde kullan�m s�ras�ndaki saklama s�releri ve kullan�m �ncesindeki ko�ullar kullan�c�n�n sorumlulu�undad�r ve normal olarak 2°C-8°C'de 24 saati a�mamal�d�r.
Ayr�ca PE/PVC inf�zyon torbas�nda seyreltilen �r�n (%0,9 sodyum klor�r veya %5'lik dekstrozun sudaki ��zeltisi i�erisinde) fiziksel ve kimyasal olarak 24 saat s�resince 2°C-8°C'de ve sonra da 12 saat s�resince 30°C alt�ndaki oda s�cakl���nda stabildir.
Kullan�lmam�� olan �r�nler ya da at�k materyaller “T�bbi At�klar�n Kontrol� Y�netmeli�i” ve “Ambalaj ve Ambalaj At�klar�n�n Kontrol� Y�netmeli�i'ne” uygun olarak imha edilmelidir.
 Pankreas Kanseri
Pankreas karn�n alt k�sm�nda yatay �ekilde bulunan bir organd�r. Sindirime yard�mc� olan enzimleri ve kan �ekerini y�netmeye yard�mc� olan hormonlar� v�cuda da��tmakla g�revlidir.
Pankreas Kanseri
Pankreas karn�n alt k�sm�nda yatay �ekilde bulunan bir organd�r. Sindirime yard�mc� olan enzimleri ve kan �ekerini y�netmeye yard�mc� olan hormonlar� v�cuda da��tmakla g�revlidir. |
 Mide Kanseri
Mide kanseri genellikle mideyi t�m�yle kaplayan ve mukus �retmekle g�revli h�crelerde ba�lar. Bu kanser tipine adenokarsinom denir.
Mide Kanseri
Mide kanseri genellikle mideyi t�m�yle kaplayan ve mukus �retmekle g�revli h�crelerde ba�lar. Bu kanser tipine adenokarsinom denir. |
�LA� E�DE�ERLER�
| E�de�er �la� Ad� | Barkodu | �la� Fiyat� |
|---|---|---|
| NOVEX | 8699514760047 | 11,712.71TL |
| REDDITUX | 8680885576016 | 5,201.10TL |
| RIXATHON | 8681428761449 | 11,712.71TL |
| RUXIENCE | 8681308771032 | 3,483.35TL |
| TRUXIMA | 8680614140143 | 11,712.71TL |
| Di�er E�de�er �la�lar |
 |
Artrit Artrit, olduk�a yayg�n bir hastal�kt�r ancak iyi anla��lamam��t�r. Asl�nda �artrit� tek bir hastal���n ad� de�ildir; eklem a�r�s� veya eklem hastal�klar�n� adland�rman�n gayri resmi yoludur. |
 |
L�semi Kan Kanseri L�semi, kan kanseridir ve v�cudunun kan olu�turan dokular�n�n hastalanmas� anlam�na gelir. Bir�ok l�semi t�r� vard�r; baz� l�semi t�rleri �ocuklarda baz�lar� da yeti�kinlerde s�k g�r�l�r. |
 |
Omurilik zedelenmeleri Omurilik zedelenmesini takip eden birka� g�n i�inde, hi�kimse hasarin ne kadar olacagini tahmin edemez. Buradaki sorun, omuriligin herhangi bir zedelenmesinden hemen sonra, bir omurilik sokunun olusmasidir. |
�LA� GENEL B�LG�LER�
CELLTRION Healthcare �la� San. ve Tic. Ltd.�ti
| Sat�� Fiyat� | 11712.71 TL [ 3 Aug 2026 ] |
| �nceki Sat�� Fiyat� | 11712.71 TL [ 27 Jul 2026 ] |
| Original / Jenerik | Original �la� |
| Re�ete Durumu | Normal Re�eteli bir ila�d�r. |
| Barkodu | 8680614140143 |
| Etkin Madde | Rituksimab |
| ATC Kodu | L01XC02 |
| Birim Miktar | 100 |
| Birim Cinsi | MG |
| Ambalaj Miktar� | 2 |
| Antineoplastik ve �mm�nomod�lat�r Ajanlar > Di�er Kanser �la�lar� > Rituksimab |
| �thal ( ref. �lke : Isvicre ) ve Be�eri bir ila�d�r. |