TENIPRA 120 mg enterik sert kapsül (14 kapsül) Kısa Ürün Bilgisi
{ Dimetil Fumarat }
1. BEŞERİ TIBBİ ÜRÜNÜN ADI
TENİPRA 120 mg gastrorezistan sert kapsül
2. KALİTATİF VE KANTİTATİF BİLEŞİM
Etkin madde
Her bir kapsül 120 mg dimetil fumarat içerir.
Yardımcı maddeler
Yardımcı maddeler için 6.1'e bakınız.
3. FARMASÖTİK FORMU
Gastrorezistan sert kapsül.
Opak beyaz gövdeli ve opak açık yeşil kapaklı sert jelatin kapsül içerisinde beyaz veya beyazımsı, enterik kaplı mini tabletler
4. KLİNİK ÖZELLİKLER
4.1. Terapötik endikasyonlar
TENİPRA, relapsing-remitting multipl sklerozu (RRMS) olan yetişkin ve 13 yaş ve üzeri pediyatrik hastaların tedavisinde endikedir.
4.2. Pozoloji ve uygulama şekli
TENİPRA tedavisi, multipl skleroz tedavisinde uzmanlaşmış bir doktorun denetimi altında başlatılmalıdır.
Pozoloji/uygulama sıklığı ve süresi
Başlangıç dozu, günde iki kez 120 mg'dir. Yedi gün sonra, günde iki kez 240 mg'lık önerilen idame doza çıkarılmalıdır. (Bölüm 4.4'e bakınız)
Eğer hasta bir dozu kaçırırsa, çift doz alınmamalıdır. Hasta kaçırılan dozu ancak dozlar arasında 4 saat bıraktığında alabilir. Aksi takdirde, hasta bir sonraki planlanan doza kadar beklemelidir.
Dozun günde iki kez 120 mg'ye geçici olarak azaltılması kızarma ve gastrointestinal advers reaksiyonların meydana gelmesini azaltabilir. Bir ay içinde, günde iki kez 240 mg'lik önerilen idame doza yeniden başlanmalıdır.
TENİPRA yiyeceklerle birlikte alınmalıdır (bkz. Bölüm 5.2). Kızarma veya gastrointestinal advers reaksiyonlar gelişen hastalarda, TENİPRA'nın yiyeceklerle birlikte alınması tolere edilebilirliğini iyileştirebilir (bkz. Bölüm 4.4, 4.5 ve 4.8).
Uygulama şekli
TENİPRA oral yoldan kullanım içindir.
Kapsül bütün olarak yutulmalıdır. Mikrotabletlerin enterik kaplaması midede tahriş edici etkileri önlemekte olduğundan; kapsül veya içeriği ezilmemeli, bölünmemeli, çözülmemeli, emilmemeli veya çiğnenmemelidir.
Özel popülasyonlara ilişkin ek bilgiler:
Böbrek/Karaciğer yetmezliği:
TENİPRA böbrek ve karaciğer yetmezliği olan hastalarda çalışılmamıştır. Klinik farmakoloji çalışmalarına dayalı olarak, hiçbir doz ayarlaması gerekmemektedir (bkz. Bölüm 5.2). Şiddetli böbrek veya şiddetli karaciğer yetmezliği olan hastaları tedavi ederken dikkatli olunmalıdır (bkz. Bölüm 4.4).
Pediyatrik popülasyon:
Pozoloji yetişkinlerde ve 13 yaş ve üzerindeki pediyatrik hastalarda aynıdır. Şu anki mevcut veriler 4.4, 4.8, 5.1 (farmakodinamik ve farmakokinetik özellikler) bölümlerinde sunulmaktadır.
10 ila 12 yaş arasındaki çocuklarda sınırlı veri mevcuttur.
10 yaşından küçük çocuklarda TENİPRA'nın güvenlilik ve etkililiği henüz belirlenmemiştir.
Geriyatrik popülasyon:
Dimetil fumarat ile yapılan klinik çalışmalar 55 yaş ve üstündeki hastalarda sınırlıdır ve bu çalışmalar, genç hastalardan farklı yanıt verip vermediğini belirlemek için yeterli sayıda 65 yaş ve üzeri hasta içermemiştir (bkz. Bölüm 5.2). Etkin maddenin etki şekline dayalı olarak, yaşlılarda doz ayarlaması gerektirecek hiçbir teorik neden yoktur.
4.3. Kontrendikasyonlar
TENİPRA, etkin maddeye veya Bölüm 6.1'de listelenen yardımcı maddelerden herhangi birine aşırı duyarlılığı bulunan kişilerde kontrendikedir.
TENİPRA, şüpheli veya doğrulanmış İlerleyici Multifokal Lökoensefalopati (PML) bulunan kişilerde kontrendikedir.
4.4. Özel kullanım uyarıları ve önlemleri
Şiddetli ve uzamış lenfopeni varlığında TENİPRA kullanan hastalarda Progresif Multifokal Lökoensefalopati (PML) vakaları meydana gelmektedir. PML'ye dair ilk belirti ve semptom görüldüğünde TENİPRA tedavisi hemen durdurulmalı ve uygun diagnostik değerlendirme yapılmalıdır (bkz. PML altbaşlığı).
Kan/Laboratuvar testleri
Dimetil fumarat ile tedavi edilen hastalarda yapılan klinik araştırmalarda, böbrek fonksiyon testlerinde değişiklikler görülmüştür (bkz. Bölüm 4.8). Bu değişikliklerin klinik önemi bilinmemektedir. Böbrek fonksiyonu (örn. kreatinin, kan üre azotu ve idrar tahlili) değerlendirmelerinin tedaviye başlamadan önce, tedavinin 3. ve 6. ayı sonunda, ardından her 6 ila 12 ayda bir ve klinik olarak gerekli görüldüğünde yapılması önerilir.
Karaciğer enzim artışı (≥3 Üst normal limit (ULN)) ve total bilirubin seviyelerinin yükselmesi (≥2 ULN) dahil ilaca bağlı karaciğer hasarı, dimetil fumarat tedavisinden kaynaklanabilmektedir. Başlangıç zamanı hemen, birkaç hafta veya daha uzun bir süre sonra olabilir. Tedavi sonrası, advers reaksiyonların çözümü gözlemlenmiştir. Klinik olarak belirtildiği gibi, tedaviye başlamadan önce ve tedavi sırasında serum aminotransferazları (örn. alanin aminotransferaz (ALT), aspartat aminotransferaz (AST)) ve total bilirubin seviyelerinin değerlendirilmesi önerilmektedir.
TENİPRA ile tedavi edilen hastalarda lenfopeni gelişebilir (bkz. Bölüm 4.8). TENİPRA ile tedaviye başlamadan önce, lenfositler de dahil olmak üzere mevcut tam kan sayımı yapılmalıdır. Lenfosit sayısının normal aralığın altında olduğu tespit edilirse TENİPRA tedavisine başlamadan önce olası nedenlerin kapsamlı bir değerlendirmesi tamamlanmalıdır. Dimetil fumarat önceden düşük lenfosit sayılarına sahip hastalarda çalışılmamıştır ve bu hastalar tedavi edilirken dikkatli olunmalıdır. TENİPRA şiddetli lenfopeni (lenfosit sayısı < 0.5 x 10/L) olan hastalarda başlatılmamalıdır. Tedaviye başladıktan sonra, her 3 ayda bir lenfosit dahil tam kan sayımı yapılmalıdır.
Lenfopeni olan hastalarda ilerleyici multifokal Lökoensefalopati (PML) riskinin artması nedeniyle aşağıdakiler için son derece dikkatli olunmalıdır:
TENİPRA, 6 aydan fazla süren uzun süreli şiddetli lenfopeni (lenfosit sayısı < 0.5 x 10/L) olan hastalarda kesilmelidir.
4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleri
Dimetil fumarat antineoplastik veya immünosüpresif tedavi kombinasyonları ile çalışılmamıştır ve dolayısıyla, eşzamanlı uygulama sırasında dikkatli olunmalıdır. Multipl skleroz klinik çalışmalarında, kısa süreli intravenöz kortikosteroidler ile nükslerin eşzamanlı tedavisi klinik açıdan anlamlı bir enfeksiyon artışı ile ilişkilendirilmemiştir.
Ulusal aşılama programlarına göre inaktif aşıların TENİPRA tedavisi sırasında eş zamanlı uygulanması düşünülebilir. Relapsing remitting multipl sklerozu olan toplam 71 hastayı kapsayan bir klinik çalışmada, en az altı aydır (n=38) günde iki kere dimetil fumarat 240 mg
ya da en az üç aydır (n=33) pegile olmayan interferon alan hastalarda, tetanoz toksoid (recall antijen) ve konjuge meningokokal C polisakkarit aşısına (neoantigen) benzer bir bağışıklık tepkisi kurulurken (aşılama titresinden sonraya kadar ≥2 kat artışı olarak tanımlanır), konjuge edilmemiş 23 valanslı pnömokokal polisakkarit aşısının (T hücresinden bağımsız antijen) farklı serotiplerine karşı bağışıklık tepkisi her iki tedavi grubunda da değişmiştir. Her iki tedavi grubunda çok az hastada, üç aşıya karşı antikor titresinde en az 4 kat ve daha fazla artış olarak tanımlanan pozitif bir immün yanıt elde edilmiştir. Tetanoz toksoidine ve pnömokok serotip 3 polisakkarite karşı yanıtta küçük sayısal farklar pegile interferon lehine not edilmiştir.
Dimetil fumarat alan hastalarda canlı atenüe aşıların etkililiği ve güvenliliği hakkında klinik veri bulunmamaktadır. Canlı aşılar artmış bir klinik enfeksiyon riski taşıyabilir ve istisnai durumlarda, bireyin aşılanmamasının oluşturacağı risk bu potansiyel riske göre ağır basmadıkça, TENİPRA ile tedavi edilen hastalara verilmemelidir.
TENİPRA tedavisi sırasında, diğer fumarik asit türevlerinin (topikal veya sistemik) birlikte kullanımından kaçınılmalıdır.
İnsanlarda, dimetil fumarat sistemik dolaşıma ulaşmadan önce esterazlar tarafından büyük ölçüde metabolize edilmektedir. Sonraki metabolizma, sitokrom P450 (CYP) sistemine dahil olmadan trikarboksilik asit siklusu üzerinden meydana gelmektedir. Potansiyel ilaç etkileşim riskleri, in vitro CYP-inhibisyon ve indüksiyon çalışmaları, bir p-glikoprotein çalışması ya da dimetil fumarat ve monometil fumaratın (dimetil fumaratın primer bir metaboliti) proteine bağlanma çalışmalarından belirlenmemiştir.
Multipl sklerozu olan hastalarda yaygın olarak kullanılan tıbbi ürünler, intramüsküler interferon beta-1a ve glatiramer asetat, dimetil fumarat ile potansiyel etkileşimler için klinik olarak test edilmiştir ve dimetil fumaratın farmakokinetik profilini değiştirmemiştir.
Sağlıklı gönüllü çalışmalarından elde edilen veriler, dimetil fumarata bağlı kızarmanın muhtemelen prostaglandin aracılı olduğunu göstermektedir. Sağlıklı gönüllülerde yapılan iki çalışmada, dimetil fumarattan 30 dakika önce, sırası ile 4 gün ve 4 hafta boyunca dozlanan 325 mg (veya eşdeğeri) enterik kaplı olmayan asetilsalisilik asitin uygulanması, dimetil fumaratın farmakokinetik profilini değiştirmemiştir. Asetilsalisilik asit tedavisi ile ilişkili potansiyel riskler, Relapsing Remitting MS hastalarında dimetil fumarat ile eşzamanlı
uygulama yapılmadan önce düşünülmelidir. Asetilsalisilik asitin uzun süreli (> 4 hafta) sürekli kullanımı araştırılmamıştır (bkz. Bölüm 4.4 ve 4.8).
Nefrotoksik tıbbi ürünler (örn. aminoglikozidler, diüretikler, nonsteroidal anti-inflamatuar ilaçlar veya lityum), TENİPRA alan hastalarda renal advers reaksiyon (örn. proteinüri bkz. Bölüm 4.8) potansiyelini arttırabilir (bkz. Bölüm 4.4 Kan/Laboratuvar testleri).
Orta miktarda alkol tüketimi dimetil fumarata maruziyeti değiştirmemiştir ve advers reaksiyonlarda bir artışla ilişkilendirilmemiştir. Alkol gastrointestinal advers reaksiyonların sıklığını arttırabileceği için, TENİPRA aldıktan sonra bir saat içinde yüksek miktarlarda sert alkollü içkilerin (alkol hacmi %30'un üzerinde olan) tüketilmesinden kaçınılmalıdır.
İn vitro CYP indüksiyon çalışmaları, dimetil fumarat ve oral kontraseptifler arasında bir etkileşim göstermemiştir. Dimetil fumarat ile kombine oral bir kontraseptifin (norgestimate ve etinil estradiol) birlikte uygulandığı in vivo bir çalışmada, oral kontraseptif kullanımı ile ilişkili herhangi bir değişiklik ortaya çıkmamıştır. Diğer progestojenleri içeren oral kontraseptiflerle herhangi bir etkileşim çalışması yapılmamıştır ancak bunların maruziyetinde dimetil fumaratın etkisi beklenmemektedir.
Özel popülasyonlara ilişkin ek bilgiler Pediyatrik popülasyon:
Etkileşim çalışmaları sadece yetişkinlerde yapılmıştır.
4.6. Gebelik ve laktasyon
Gebelik kategorisi: C
Çocuk doğurma potansiyeli bulunan kadınlar/Doğum kontrolü (Kontrasepsiyon) TENİPRA'nın çocuk doğurma potansiyeli bulunan kadınlarda kullanımına ilişkin yeterli veri mevcut değildir.
TENİPRA gebelik sırasında ve uygun kontrasepsiyon yöntemlerini kullanmayan çocuk doğurma potansiyeline sahip kadınlarda önerilmemektedir (bkz. Bölüm 4.5).
Gebelik dönemi:
Hayvanlar üzerinde yapılan araştırmalar üreme toksisitesinin bulunduğunu göstermiştir (bkz. Bölüm 5.3). İnsanlara yönelik potansiyel risk bilinmemektedir.
Dimetil fumaratın gebe kadınlarda kullanımına ilişkin yeterli veri mevcut değildir.
TENİPRA gebelikte yalnızca gerekli olduğunda ve beklenen yararın fetüse olan potansiyel zarardan daha fazla olması durumunda kullanılmalıdır.
TENİPRA gerekli olmadıkça gebelik döneminde kullanılmamalıdır.
Laktasyon dönemi:
Dimetil fumaratın veya metabolitlerinin insan sütü ile atılıp atılmadığı bilinmemektedir. Yeni doğanlar/infantlar için risk göz ardı edilemez. Emzirmenin durdurulup durdurulmayacağına ya da TENİPRA tedavisinin durdurulup durdurulmayacağına/tedaviden kaçınılıp kaçınılmayacağına ilişkin karar verilirken, emzirmenin çocuk açısından faydası ve TENİPRA tedavisinin emziren anne açısından faydası dikkate alınmalıdır.
Üreme yeteneği / Fertilite:
Dimetil fumaratın insanlarda fertilite üzerine etkilerine dair hiçbir veri bulunmamaktadır. Klinik öncesi çalışmalardan elde edilen veriler, dimetil fumaratın fertiliteyi azaltma riskinin olduğunu düşündürmemektedir (bkz. Bölüm 5.3).
4.7. Araç ve makine kullanımı üzerindeki etkiler
TENİPRA'nın araç ve makine kullanma yetisi üzerinde etkisi yoktur veya ihmal edilebilir düzeydedir. TENİPRA'nın araç ve makine kullanma yetisi üzerine hiçbir çalışma yürütülmemiştir ancak klinik çalışmalarda bu yetiye potansiyel etkisi olan dimetil fumarat ile ilişkili hiçbir etki gözlenmemiştir.
4.8. İstenmeyen etkiler
Güvenlilik profilinin özeti
Dimetil fumarat ile tedavi edilen hastalarda en yaygın advers reaksiyonlar (insidans ≥%10); kızarma ve gastrointestinal rahatsızlıklardır (ishal, bulantı, karın ağrısı, üst karın ağrısı). Kızarma ve gastrointestinal rahatsızlıklar, tedavinin erken dönemlerinde başlama eğilimi gösterir (en çok ilk bir ay içinde). Kızarma ve gastrointestinal rahatsızlıklar yaşayan hastalarda, bu olaylar TENİPRA tedavisi boyunca aralıklarla meydana gelmeye devam edebilir. Dimetil fumarat ile tedavi edilen hastalarda tedavinin kesilmesine (insidans >%1) yol
açan en yaygın olarak bildirilen advers reaksiyonlar kızarma (%3) ve gastrointestinal olaylardır (%4).
Plasebo-kontrollü ve kontrolsüz klinik çalışmalarda, toplam 2513 hasta TENİPRA'yı 12 yıla kadar olan süreler boyunca kullanmıştır ve toplam maruziyet 11.318 kişi yılına eşdeğer olmuştur. Toplam 1.169 hasta TENİPRA ile en az 5 yıl ve 426 hasta en az 10 yıl tedavi görmüştür. Kontrolsüz klinik çalışmalardan elde edilen deneyimler, plasebo kontrollü klinik çalışmalardaki deneyimler ile uyumlu bulunmuştur.
Klinik çalışmalardan kaynaklanan advers reaksiyonlar, ruhsatlandırma sonrası güvenlik ve spontan raporlar aşağıdaki tabloda sunulmaktadır.
Advers reaksiyonlar, MedDRA Sistem Organ Sınıfı altında MedDRA tercih edilen terimlere göre belirtilmiştir. Advers reaksiyonların insidansı aşağıdaki kategorilere göre ifade edilmektedir:
Çok yaygın (≥ 1/10)
- Yaygın (≥1/100 ila <1/10)
4.9. Doz aşımı ve tedavisi
Dimetil fumarat ile doz aşımı vakaları bildirilmiştir. Bu vakalarda tanımlanan semptomlar, TENİPRA'nın bilinen advers reaksiyon profili ile uyumludur. TENİPRA'nın eliminasyonunu arttırmak için bilinen terapötik bir müdahale yoktur ve bilinen bir antidot bulunmamaktadır. Doz aşımı durumunda, klinik olarak belirtildiği gibi semptomatik destekleyici tedaviye başlanması önerilmektedir.
5. FARMAKOLOJİK ÖZELLİKLER
5.1. Farmakodinamik özellikler
Farmakoterapötik grup: Diğer immünosüpresanlar ATC kodu: L04AX07Etki mekanizması
Multipl sklerozda, dimetil fumarat tarafından sergilenen terapötik etki mekanizması tam olarak anlaşılamamıştır. Klinik öncesi çalışmalar, dimetil fumaratın farmakodinamik yanıtlarının esas olarak Nükleer faktör (eritroid-derived 2)-like 2 (Nrf2) transkripsiyon yolağının aktivasyonu aracılığıyla gerçekleştiğini göstermektedir. Dimetil fumaratın, hastalarda Nrf2-bağımlı antioksidan genlerini regüle ettiği gösterilmiştir (örn. NAD(P)H dehidrojenaz, kinon 1; [NQO1]).
Farmakodinamik etkiler
İmmün sistem üzerine etkiler
Klinik öncesi ve klinik çalışmalarda, dimetil fumarat anti-inflamatuar ve immünomodülatör özellikler göstermiştir. Dimetil fumarat ve dimetil fumaratın primer metaboliti monometil fumarat, klinik öncesi modellerde inflamatuvar uyaranlara yanıt olarak immün hücre aktivasyonunu ve sonrasında meydana gelen pro-inflamatuvar sitokinlerin salınımını azaltmıştır. Psöriyazisi olan hastalarda yapılan klinik çalışmalarda, dimetil fumarat pro inflamatuvar sitokin profillerinin (T1, T17) down-regülasyonu ile lenfosit fenotiplerini etkilemiş ve anti-inflamatuvar üretimine (T2) eğilim göstermiştir. Dimetil fumarat, multiple inflamatuvar ve nöroinflamatuvar hasar modelinde terapötik aktivite göstermiştir. MS hastalarda Faz 3 çalışmalarda, (DEFINE, CONFIRM ve ENDORSE), TECFIDERA ile tedavi, ortalama lenfosit sayımlarından sonra ortalama bir plato göstermiştir; bu, ilk yıl boyunca
başlangıç değerlerinin yaklaşık %30'u kadar azalmış ve ardından bir plato izlemiştir. Bu çalışmalarda, lenfosit sayıları normalin alt sınırının (LLN, 910 hücre/mm3) altındayken TENİPRA tedavisini bırakan hastalar, lenfosit sayılarının LLN'ye geri kazanılması için izlenmiştir.
Şekil 1, uzamış şiddetli lenfopeni olmaksızın Kaplan-Meier yöntemine dayalı olarak LLN'ye ulaştığı tahmin edilen hastaların oranını göstermektedir. İyileşme başlangıç çizgisi (RBL), TENİPRA'nın kesilmesinden önceki tedavideki son ALC olarak tanımlandı. RBL'de hafif, orta veya şiddetli lenfopenisi olan 12. Hafta ve 24. Haftada LLN'ye (ALC ≥ 0.9 x 109//L) iyileşen hastaların tahmini oranı, %95 noktasal güven aralıklarıyla Tablo 1, Tablo 2 ve Tablo 3'te sunulmaktadır. Hayatta kalma fonksiyonunun Kaplan-Meier tahmin edicisinin standart hatası Greenwood formülü kullanılarak hesaplanır.
Şekil 1: Kaplan-Meier Metodu; İyileşme Başlangıç Noktasından (RBL) ≥ 910 hücre/mm 3 LLN'ye İyileşen Hastaların Oranı
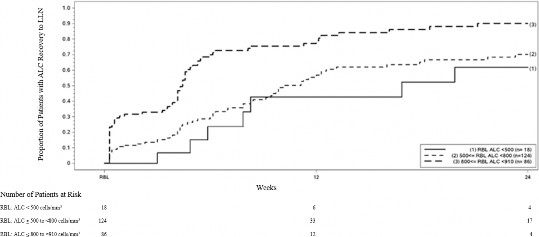
Tablo 1: Kaplan-Meier Yöntemi; Uzamış şiddetli lenfopenisi olan hastalar hariç, LLN'ye ulaştığı tahmin edilen hastaların oranı, iyileşme başlangıcında (RBL) hafif lenfopeni
Risk altundaki hafif lenfopenisi olan hasta sayısı | Taban Çizgisi N=86 | Hafta 12 N=12 | Hafta 24 N=4 |
LLN'ye ulaşan oran (0,95 GA) |
| 0.81 (0.71, 0.89) | 0.90 (0.81, 0.96) |
Tablo 2: Kaplan-Meier Yöntemi; Uzamış şiddetli lenfopenisi olan hastalar hariç, LLN'ye ulaştığı tahmin edilen hastaların oranı, iyileşme başlangıcında (RBL) orta derecede lenfopeni
Risk altundaki orta derecede lenfopenisi olan hasta sayısı | Taban Çizgisi N=124 | Hafta 12 N=33 | Hafta 24 N=17 |
LLN'ye ulaşan oran (0,95 GA) |
| 0.57 (0.46, 0.67) | 0.70 (0.60, 0.80) |
Tablo 3: Kaplan-Meier Yöntemi; Uzamış şiddetli lenfopenisi olan hastalar hariç, LLN'ye ulaştığı tahmin edilen hastaların oranı, iyileşme başlangıcında (RBL) şiddetli lenfopeni
Risk altundaki ciddi derecede lenfopenisi olan hasta sayısı | Taban Çizgisi N=18 | Hafta 12 N=6 | Hafta 24 N=4 |
LLN'ye ulaşan oran (0,95 GA) |
| 0.43 (0.20, 0.75) | 0.62 (0.35, 0.88) |
Klinik etkililik ve güvenlilik
Relapsing-remitting multipl sklerozlu (RRMS) hastalar ile iki adet, 2 yıllık, randomize, çift kör, plasebo kontrollü çalışmalar [1234 hasta ile (DEFINE) ve 1417 hasta ile (CONFIRM)] yapılmıştır. MS'in ilerleyici formları olan hastalar bu çalışmalara dahil edilmemiştir. Etkililik (bkz. aşağıdaki tablo) ve güvenlilik, randomizasyondan önceki bir yıllık süreçte en az 1 nüks yaşayan hastalarda veya randomizasyondan önce altı haftada en az bir gadolinyum-tutan (Gd+) lezyon geliştirerek bir beyin Manyetik Rezonans Görüntülemesi (MRG) yapılan hastaları kapsayan 0'dan 5'e değişen Genişletilmiş Engellilik Durum Ölçeği (EDSS) skorlu hastalarda kanıtlanmıştır. CONFIRM, glatiramer asetatın referans karşılaştırıcısı olarak değerlendirici bir kör (örn. tedavi çalışmasının sonuçlarını değerlendiren çalışma hekimi/araştırmacı körleştirilmiştir) içermiştir.
DEFINE'da, hastalar aşağıdaki medyan başlangıç özelliklerine sahipti: yaş 39, hastalık süresi
7.0 yıl, EDSS skoru 2.0. Ek olarak, hastaların %16'sının EDSS puanı >3.5'dir ve %28'i önceki yıl en az 2 nüks yaşamış ve %42'si halihazırda onaylanmış başka MS tedavileri almıştır. MRG kohortunda, çalışmaya giren hastaların %36'sında başlangıçta Gd+ lezyonları mevcuttur (ortalama Gd+ lezyon sayısı 1,4).
CONFIRM'de, hastalar şu özellikleri sergilemiştir: yaş 37, hastalık süresi 6,0 yıl, EDSS skoru 2,5. Buna ek olarak, hastaların %17'si EDSS skoru >3,5'dir ve %32'si önceki yıl en az 2 nüks geçirmiş ve %30'u önceden başka onaylı MS tedavileri almıştır. MRG kohortunda, çalışmaya giren hastaların %45'inde başlangıçta Gd+ lezyonları mevcuttur (ortalama Gd+ lezyon sayısı 2,4).
Plaseboyla karşılaştırıldığında, dimetil fumarat ile tedavi edilen hastalarda şu değerlerde klinik açıdan anlamlı ve istatistiksel açıdan önemli bir azalma gözlenmiştir: DEFINE'da primer sonlanım noktası, 2 yılda nüks yapan hasta oranı; ve Çalışma 2'de primer sonlanım noktası, 2 yılda yıllık nüks oranı (ARR).
CONFIRM'de; glatiramer asetat ve plasebo için ARR, onaylanmış kısa ürün bilgisi ve kullanma talimatına uygun olarak %29'luk bir azalmaya karşılık gelecek şekilde, sırasıyla 0,286 ve 0,401 olarak belirlenmiştir.
| DEFINE | CONFIRM | |||
|
Plasebo | Dimetil fumarat günde iki kez 240 mg |
Plasebo | Dimetil fumarat günde iki kez 240 mg | Glatiramer asetat |
Klinik Sonlanım Noktaları | |||||
Hasta sayısı | 408 | 410 | 363 | 359 | 350 |
Yıllık nüks oranı | 0,364 | 0,172 | 0,401 | 0,224 | 0,286 |
İnsidans yoğunluk oranı (%95 GA) |
| 0,47 (0,37, 0,61) |
| 0,56 (0,42, 0,74) | 0,71 (0,55, 0,93) |
Nüks oranı | 0,461 | 0,270 | 0,410 | 0,291 | 0,321 |
Risk oranı (%95 GA) |
| 0,51 (0,40, 0,66) |
| 0,66 (0,51, 0,86) | 0,71 (0,55, 0,92) |
12 haftalık doğrulanmış engellilik progresyon oranı | 0,271 | 0,164 | 0,169 | 0,128 | 0,156 |
Risk oranı (%95 GA) |
| 0,62 (0,44, 0,87) |
| 0,79 (0,52, 1,19) | 0,93 (0,63, 1,37) |
24 haftalık doğrulanmış engellilik progresyon oranı | 0,169 | 0,128 | 0,125 | 0,078 | 0,108 |
Risk oranı (%95 GA) |
| 0,77 (0,52, 1,14) |
| 0,62 (0,37, 1,03) | 0,87 (0,55, 1,38) |
MRI Sonlanım Noktaları | |||||
Hasta sayısı | 165 | 152 | 144 | 147 | 161 |
2 yılda yeni gelişen veya gelişmekte olan T2 lezyonlarının ortalama (medyan) sayısı |
16,5 (7,0) |
3,2 (1,0) |
19,9 (11,0) |
5,7 (2,0) |
9,6 (3,0) |
Ortalama lezyon oranı (%95 GA) |
| 0,15 (0,10, 0,23) |
| 0,29 (0,21, 0,41) | 0,46 (0,33, 0,63) |
2 yılda Gd lezyonların ortalama (medyan) sayısı | 1,8 (0) | 0,1 (0) | 2,0 (0,0) | 0,5 (0,0) | 0,7 (0,0) |
Risk oranı (%95 GA) |
| 0,10 (0,05, 0,22) |
| 0,26 (0,15, 0,46) | 0,39 (0,24, 0,65) |
2 yılda yeni T1 hipointens lezyonlarının ortalama (medyan) sayısı | 5,7 (2,0) | 2,0 (1,0) | 8,1 (4,0) | 3,8 (1,0) | 4,5 (2,0) |
Lezyon ortalama oranı (%95 GA) |
| 0,28 (0,20, 0,39) |
| 0,43 (0,30, 0,61) | 0,59 (0,42, 0,82) |
Kontrolsüz 8 yıllık bir uzatma çalışmasına (ENDORSE), pivot çalışmalardan (DEFINE ve CONFIRM) 1.736 uygun RRMS hastasını kaydetmiştir. Çalışmanın birincil amacı, RRMS'li hastalarda TENİPRA'nın uzun vadeli güvenliğini değerlendirmekti. 1.736 hastanın yaklaşık yarısı (909, %52) 6 yıl veya daha uzun süre tedavi görmüştür. 3 çalışmanın tamamında 501 hasta sürekli olarak günde iki kez 240 mg TENİPRA ile tedavi edildi ve daha önce DEFINE ve CONFIRM çalışmalarında plasebo ile tedavi edilen 249 hasta, ENDORSE çalışmasında günde iki kez 240 mg tedavi aldı.Sürekli olarak günde iki kez tedavi alan hastalar 12 yıla kadar tedavi edildi.
ENDORSE çalışması sırasında, günde iki kez 240 mg TENİPRA ile tedavi edilen tüm hastaların yarısından fazlasında nüks olmadı. 3 çalışmanın tamamında sürekli olarak günde iki kez tedavi edilen hastalar için, ayarlanmış ARR, DEFINE ve CONFIRM çalışmalarında
0.187 (95% GA: 0.156, 0.224) ve ENDORSE çalışmasında 0.141 (95% GA: 0.119, 0.167)
'dir. Daha önce plasebo ile tedavi edilen hastalar için, DEFINE ve CONFIRM çalışmalarında
0,330 olan düzeltilmiş ARR (95% GA: 0.266, 0.408), ENDORSE çalışmasında 0,149 (95% GA: 0.116, 0.190)'a düşmüştür.
ENDORSE çalışmasında, hastaların çoğu (> %75) sakatlığın ilerlemesini doğrulamamıştır (6 aylık kalıcı sakatlık ilerlemesi olarak ölçülmüştür). Üç çalışmadan elde edilen havuzlanmış sonuçlar, TENİPRA ile tedavi edilen hastaların, ENDORSE genelinde ortalama EDSS puanlarında hafif bir artış ile tutarlı ve düşük doğrulanmış engellilik ilerleme oranlarına sahip olduğunu göstermiştir. MRI değerlendirmeleri (6 yıla kadar, daha önce DEFINE ve CONFIRM çalışmalarının MRI kohortuna dahil edilmiş 752 hasta dahil olmak üzere) hastaların çoğunluğunda (yaklaşık %90) Gd'yi artıran lezyonları olmadığını göstermiştir. 6 yıl boyunca, yeni veya yeni genişleyen T2 ve yeni T1 lezyonlarının yıllık düzeltilmiş ortalama sayısı düşük kalmıştır.
Yüksek hastalık aktivitesi gösteren hastalarda etkililik
3 aylık uzamış engellilik progresyonuna kadar geçen zamana etkisi net bir şekilde belirlenmemişken, yüksek hastalık aktivitesine sahip bir hasta alt grubunda nüksler üzerine uygun bir tedavi etkisi gözlenmiştir. Çalışmaların dizaynı nedeni ile, yüksek hastalık aktivitesi aşağıdaki şekilde tanımlanmıştır:
5.2. Farmakokinetik özellikler
Oral olarak uygulanan dimetil fumarat esterazlar tarafından presistemik olarak hızlıca hidrolize uğrar ve aktif olan monometil fumarat adlı primer metabolitine dönüştürülür. Dimetil fumarat, TENİPRA'nın oral olarak alınmasından sonra plazmada ölçülemez. Bu nedenle dimetil fumarat ile ilgili tüm farmakokinetik analizler plazma monometil fumarat konsantrasyonlarıyla gerçekleştirilmiştir. Farmakokinetik veriler, multipl sklerozlu ve sağlıklı gönüllülerden oluşan deneklerden elde edilmiştir.
Emilim:
Monometil fumaratın Tdeğeri 2 ila 2,5 saattir. Dimetil fumarat, enterik sert kapsül, enterik bir kaplama tarafından korunan mikrotabletler içerdiğinden, emilim kapsüller mideyi terk edene kadar başlamaz (genellikle 1 saatten az). Günde iki kez yiyeceklerle birlikte alınan 240 mg'ın ardından, multipl sklerozlu deneklerde medyan pik (C) 1,72 mg/L ve genel eğri altındaki alan (EAA) maruziyet 8,02 saat.mg/L olmuştur. Genel olarak, çalışılan doz aralığında (120 mg ila 360 mg) Cve EAA, yaklaşık olarak dozla orantılı şekilde artmıştır. Multipl sklerozlu hastalarda, iki 240 mg'lık doz bir günlük doz rejimi olarak 4 saat arayla günde üç kez uygulanmıştır. Bu durum, hiçbir güvenlik önlemi olmaksızın günde iki kez uygulanan dozlamaya kıyasla %12'lik medyan C'ında bir artış sağlayarak maruziyetin minimum birikimi ile sonuçlanmıştır (günde üç kez yapılan uygulama için 1,93 mg/L'ye karşı, günde iki kez yapılan uygulama için 1,72 mg/L).
Yiyecek, dimetil fumaratın maruziyeti üzerine klinik açıdan bir etki göstermemektedir. Bununla birlikte, dimetil fumarat kızarma veya gastrointestinal advers olaylar ile ilgili gelişmiş tolere edilebilirlik nedeni ile yiyeceklerle birlikte alınmalıdır (bkz. Bölüm 4.2).
Dağılım:
240 mg dimetil fumaratın oral yolla alınmasının ardından görülen dağılım hacmi 60 L ve 90 L arasında değişmektedir. Monometil fumaratın insan plazma proteinine bağlanma oranı genelde %27 ile %40 arasındadır.
Biyotransformasyon:
İnsanlarda, dimetil fumarat büyük ölçüde metabolize olup %0,1'den azı idrarda değişmemiş olarak atılmaktadır. Dimetil fumarat, sistemik dolaşıma ulaşmadan önce ilk olarak gastrointestinal kanal, kan ve dokularda bulunan esterazlarla metabolize olur. Başka bir metabolizma, sitokrom P450 (CYP) sisteminin dahil olmadığı, trikarboksilik asit döngüsü aracılığı ile meydana gelmektedir. Tek bir 240 mg C-dimetil fumarat dozu ile yapılan çalışmada, glukoz insan plazmasındaki baskın metabolit olarak tanımlanmıştır. Dolaşımdaki diğer metabolitleri fumarik asit, sitrik asit ve monometil fumarattır. Fumarik asitin daha sonra metabolizması trikarboksilik asitten CO'in uzaklaştırılması ile olur ve bu eliminasyonun primer yoludur.
Eliminasyon:
COuzaklaştırılması, dozun %60'ının eliminasyonundan sorumlu olan primer dimetil fumarat eliminasyon yoludur. Renal ve fekal eliminasyon, sırasıyla dozun %15,5'i ve %0,9'unu elimine eden sekonder eliminasyon yollarıdır.
Monometil fumaratın terminal yarı ömrü kısadır (yaklaşık 1 saat) ve monometil fumarat bireylerin çoğunda 24 saatte dolaşımda kalmaz. Terapötik rejimde çoklu dimetil fumarat dozları ile ana ilaç veya monometil fumarat birikimi meydana gelmez.
Doğrusallık / Doğrusal olmayan durum:
Tek ve çoklu 120 mg ve 360 mg'lık doz aralığında çalışılmış olan dimetil fumaratın maruziyeti, dozla orantılı bir biçimde artmaktadır.
Özel hasta gruplarındaki farmakokinetik özellikler
Varyans Analizi (ANOVA) bulgularına dayalı olarak, Relapsing Remitting Multipl Skleroz (RRMS) hastalarında maruziyetin ana orta değişkeni (Cve EAA'ya göre) vücut ağırlığıdır. Ancak klinik çalışmalarda değerlendirilen güvenlilik ve etkililik ölçümlerini etkilememiştir.
Cinsiyet ve yaş, dimetil fumaratın farmakokinetik özellikleri üzerine klinik açıdan önemli bir etki göstermemiştir. 65 yaş ve üzeri hastalarda farmakokinetik özellikler çalışılmamıştır.
Pediyatrik popülasyon
Günde iki kez 240 mg kullanılan dimetil fumaratın farmakokinetik profili, RRMS'li 13-17 yaş arası pediyatrik hastada yapılan küçük, açık uçlu, kontrolsüz bir çalışmada değerlendirilmiştir (n=21). Dimetil fumaratın bu adolesan hastalardaki farmakokinetiği daha öncesinde yetişkin hastalarda gözlenenler ile tutarlıdır. (C: 2,00±1,29 mg/l; EAA: 3,62±1,16 sa.mg/l, tam bir günlük EAA: 7,24 sa.mg/l'ye denk gelen).
Böbrek yetmezliği
Renal yolak, dimetil fumarat için uygulanan dozun %16'sından azının atıldığı sekonder bir eliminasyon yolu olduğundan, böbrek yetmezliği olan bireylerde farmakokinetik özellikleri değerlendirilmemiştir.
Karaciğer yetmezliği
Dimetil fumarat ve monometil fumarat, CYP450 sistemi dahil olmadan esterazlar tarafından metabolize edildiğinden, karaciğer bozukluğu olan bireylerde farmakokinetik özellik değerlendirmesi yürütülmemiştir.
5.3. Klinik öncesi güvenlilik verileri
Aşağıdaki toksikoloji ve üreme toksisitesi bölümlerinde açıklanan advers reaksiyonlar, klinik çalışmalarda gözlenmemiştir. Ancak klinik maruziyet düzeylerine benzer maruziyet düzeylerinde hayvanlarda görülmüştür.
Mutajenezis:
Dimetil fumarat ve monometil fumarat bir seri in vitro testte negatif sonuç vermiştir (Ames, memeli hücrelerinde kromozomal sapma). Dimetil fumarat sıçanlarda in vivo mikroçekirdek testinde negatif sonuç vermiştir.
Karsinojenezis:
Dimetil fumaratın karsinojenite çalışmaları, fare ve sıçanlarda 2 yıla varan bir süre boyunca gerçekleştirilmiştir. Dimetil fumarat farelere 25, 75, 200 ve 400 mg/kg/gün ve sıçanlara 25, 50, 100 ve 150 mg/kg/gün'lük dozlarda oral yolla uygulanmıştır. Farelerde, önerilen insan dozuna eşdeğer maruziyette (EAA) 75 mg/kg/gün dozunda renal tübüler karsinom insidansı artmıştır. Sıçanlarda, renal tübüler karsinom ve testiküler Leydig hücre adenomu insidansı, önerilen insan dozundan yaklaşık 2 kat daha yüksek, 100 mg/kg/gün'de artmıştır. Bu bulguların insandaki risk ile ilişkisi bilinmemektedir.
Farelerde, önerilen insan dozu ile eşdeğer maruziyette ve sıçanlarda önerilen insan dozu maruziyetinin altında, nonglandüler midede (ön mide) skuamöz hücre papilloma ve karsinomu insidansı artmıştır (EAA baz alınarak). Kemirgenlerdeki ön midenin insanlarda bir karşılığı yoktur.
Toksikoloji:
Kemirgenlerde, tavşanlarda ve maymunlarda oral gavaj yoluyla uygulanan dimetil fumarat süspansiyonu (%0,8 hidroksipropil metilselülozda dimetil fumarat) ile klinik dışı çalışmalar yürütülmüştür. Kronik köpek çalışması, dimetil fumarat kapsülün oral yolla uygulanmasıyla gerçekleştirilmiştir.
Farelerde, sıçanlarda, köpeklerde ve maymunlarda dimetil fumaratın tekrarlı şekilde oral alınmasından sonra böbrek üzerinde etkiler gözlenmiştir. Hasarı düşündüren renal tübül epitelyum rejenerasyonu tüm türlerde gözlenmiştir. Renal tübüler hiperplazisi yaşam boyu dozlama ile (2 yıllık çalışma) sıçanlarda gözlenmiştir. 11 ay boyunca dimetil fumaratın günlük oral dozlarını alan köpeklerde kortikal atrofi için hesaplanan marj, EAA'ya dayalı olarak önerilen dozun 3 katında gözlenmiştir. 12 ay boyunca dimetil fumaratın günlük oral dozlarını alan maymunlarda tek hücre nekrozu, EAA'ya dayalı olarak önerilen dozun 2 katında gözlenmiştir. İnterstisyel fibrozis ve kortikal atrofi, EAA'ya dayalı olarak önerilen dozun 6 katında gözlenmiştir. Bu bulguların insanlarla ilişkisi bilinmemektedir.
Testislerdeki seminiferöz epitelin dejenerasyonu sıçanlarda ve köpeklerde görülmüştür. Bulgular sıçanlarda yaklaşık olarak önerilen dozda ve köpeklerde önerilen dozun 3 katında (EAA'ya dayalı olarak) gözlenmiştir. Bu bulguların insanlarla ilişkisi bilinmemektedir.
3 ay ya da daha uzun süreli çalışmalarda, farelerin ve sıçanların ön midesinde skuamöz epitel hiperplazisi ve hiperkeratozu; inflamasyonu ve skuamöz hücre papilloma ve karsinomu olduğu gözlenmiştir. Fare ve sıçanlardaki ön midenin insanlarda bir karşılığı yoktur.
Üreme toksisitesi:
Çiftleşme öncesinde veya sırasında 75, 250 ve 375 mg/kg/gün'de erkek sıçanlara oral dimetil fumarat uygulaması, test edilen en yüksek doza kadar (EAA'ya dayalı olarak önerilen dozun en az 2 katı) erkek fertilitesi üzerine etki göstermemiştir. Çiftleşme öncesinde ve sırasında ve gebeliğin 7. gününe kadar devam eden 25, 100 ve 250 mg/kg/gün'de dişi sıçanlara oral dimetil fumarat uygulaması, 14 günlük östrus evresi sayısını azaltmış ve test edilen en yüksek dozda (EAA'ya dayalı olarak önerilen dozun 11 katı) uzamış diöstrus gözlenen hayvan sayısını
arttırmıştır. Bununla birlikte, bu değişimler fertiliteyi veya oluşan canlı fetüs sayısını etkilememiştir.
Sırasıyla 0,48 ila 0,64 ve 0,1'lik fetal/maternal plazma konsantrasyonu oranları ile dimetil fumaratın sıçanlarda ve tavşanlarda plasenta membranından geçerek fetal kana karıştığı gösterilmiştir. Sıçanlarda veya tavşanlarda uygulanan herhangi bir dimetil fumarat dozunda hiçbir malformasyon gözlenmemiştir. Organogenez dönemi boyunca gebe sıçanlara 25, 100 ve 250 mg/kg/gün'lük oral dozlarda yapılan dimetil fumarat uygulaması EAA'ya dayalı olarak önerilen dozun 4 katında maternal advers etkilere yol açmış ve EAA'ya dayalı olarak önerilen dozun 11 katında düşük fetüs ağırlığına ve gecikmiş osifikasyona (metatarsallar ve arka bacak falenksleri) neden olmuştur. Daha düşük fetüs ağırlığı ve gecikmiş osifikasyon maternal toksisiteye (azalmış vücut ağırlığı ve gıda tüketimi) sekonder kabul edilmiştir.
Organogenez sırasında gebe tavşanlara 25, 75 ve 150 mg/kg/gün'lük oral dozlarda yapılan dimetil fumarat uygulaması, embriyo-fetal gelişim üzerine etki göstermemiştir. EAA'ya dayalı olarak önerilen dozun 7 katında azalmış maternal vücut ağırlığına neden olmuş ve önerilen dozun 16 katında da düşük artışına yol açmıştır.
4.6. Gebelik ve laktasyon
Jüvenil sıçanlarda, doğum sonrası (PND) 28. günden PND 90 ila 93'e kadar (insanlarda yaklaşık 3 yaş ve üzerindekilere eşdeğer) günlük oral dimetil fumarat uygulaması ile yapılan iki toksisite çalışması, yetişkin hayvanlarda gözlenene benzer şekilde böbrek ve ön midede benzer hedef organ toksisitelerini ortaya çıkarmıştır. İlk çalışmada, dimetil fumarat, 140 mg/kg/günlük en yüksek doza kadar gelişimi, nörodavranışı veya erkek ve kadın doğurganlığını etkilememiştir (pediyatrik hastalarda sınırlı EAA verilerine göre önerilen insan dozunun yaklaşık 4.6 katı). Benzer şekilde, erkek jüvenil sıçanlarda yapılan ikinci çalışmada (önerilen pediyatrik dozda varsayılan EAA'nın yaklaşık 15 katı) en yüksek 375 mg/kg/gün dimetil fumarat dozuna kadar erkek üreme ve aksesuar organları üzerinde hiçbir etki gözlenmemiştir. Bununla birlikte, erkek jüvenil sıçanlarda femur ve lomber vertebralarda azalmış kemik mineral içeriği ve yoğunluğu belirgindi. İn vivo olarak aynı aktif metabolit
monometil fumarata metabolize olan başka bir fumarik ester olan oral diroksimel fumarat uygulamasını takiben jüvenil sıçanlarda kemik dansitometrisi değişiklikleri de gözlenmiştir. Jüvenil sıçanlarda dansitometri değişiklikleri için NOAEL, önerilen pediatrik dozda varsayılan EAA'nın yaklaşık 1.5 katıdır. Kemik etkilerinin daha düşük vücut ağırlığı ile ilişkisi mümkündür, ancak doğrudan bir etkinin dahil edilmesi göz ardı edilemez. Kemik bulguları yetişkin hastalar için sınırlı bir öneme sahiptir. Pediyatrik hastalar için alaka düzeyi bilinmemektedir.
6. FARMASÖTİK ÖZELLİKLER
6.1. Yardımcı maddelerin listesi
Mikrokristalin selüloz Kroskarmelloz sodyumSilikanlandırılmış mikrokristalin selüloz Kolloidal silikon dioksit
Magnezyum stearat
Metakrilik asit – Metil metakrilat kopolimer (1:1) Trietil sitrat
Talk
Metakrilik asit – Etil akrilat kopolimer (1:1) dispersiyon %30 Kapsül içeriği:
Jelatin (sığır jelatini) Titanyum dioksit
Parlak mavi FCF-FD&C Blue 1 Siyah demir oksit
Sarı demir oksit
6.2. Geçimsizlikler
Uygulanabilir değildir.
6.3. Raf ömrü
24 ay
6.4. Saklamaya yönelik özel tedbirler
25C altındaki oda sıcaklığında saklayınız.
6.5. Ambalajın niteliği ve içeriği
TENİPRA 120 mg Gastrorezistan Sert Kapsül, 14 kapsül PVC/PE/PVDC – Aluminyum blister ambalajlarda kullanma talimatı ile birlikte karton kutuda piyasaya sunulmaktadır.
6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemler
Özel bir gereklilik bulunmamaktadır.
Kullanılmamış olan ürünler ya da atık materyaller "Tıbbi Atıkların Kontrolü Yönetmeliği" ve "Ambalaj Atıklarının Kontrolü Yönetmelikleri"ne uygun olarak imha edilmelidir.
 Dış Gebelik
Dış gebelik, her 100 gebelikten birini etkileyen, sık görülen ve ölüme sebep
olabilecek bir durumdur. Bu, döllenen yumurta, rahimin dışına yerleşirse, oluşan
bir durumdur. Gebelik ilerledikçe, ağrıya ve kanamalara sebep olur.
Dış Gebelik
Dış gebelik, her 100 gebelikten birini etkileyen, sık görülen ve ölüme sebep
olabilecek bir durumdur. Bu, döllenen yumurta, rahimin dışına yerleşirse, oluşan
bir durumdur. Gebelik ilerledikçe, ağrıya ve kanamalara sebep olur. |
 Ağız Kanseri
Ağız kanserinin en yaygın türleri, dudak, dil, dişetidir. Nadiren
yanak içi veya damak bölgelerini de içine alır.
Ağız Kanseri
Ağız kanserinin en yaygın türleri, dudak, dil, dişetidir. Nadiren
yanak içi veya damak bölgelerini de içine alır. |
İLAÇ EŞDEĞERLERİ
| Eşdeğer İlaç Adı | Barkodu | İlaç Fiyatı |
|---|---|---|
| DIMERAST | 8680698160358 | |
| FUMATIL | 8699828150602 | 1,543.78TL |
| LIDWINA | 8699543160078 | 1,541.23TL |
| PHARON | 8699536150796 | 1,546.28TL |
| TECDIMEX | 8680698160501 | 1,874.00TL |
| Diğer Eşdeğer İlaçlar |
 |
Diyabet Hastalığı Diyabet, insülin hormonu ile ilgili problemlerden kaynaklanan bir hastalıktır. |
 |
Mide Kanseri Mide kanseri genellikle mideyi tümüyle kaplayan ve mukus üretmekle görevli hücrelerde başlar. Bu kanser tipine adenokarsinom denir. |
 |
İnme İnme, beynin hasar görmesinin sonucudur. Bu hasar, beynin bir kısmındaki ya bir kanama ya da akut kan eksikliği nedeniyle o kısmın geçici ya da kalıcı olarak işlevini yapamamasına yol açar. |
İLAÇ GENEL BİLGİLERİ
Abdi İbrahim İlaç Sanayi ve Ticaret A.Ş.
| Satış Fiyatı | 1541.23 TL [ 3 Aug 2026 ] |
| Önceki Satış Fiyatı | 1541.23 TL [ 27 Jul 2026 ] |
| Original / Jenerik | Original İlaç |
| Reçete Durumu | Normal Reçeteli bir ilaçdır. |
| Barkodu | 8699514160014 |
| Etkin Madde | Dimetil Fumarat |
| ATC Kodu | L04AX07 |
| Birim Miktar | 120 |
| Birim Cinsi | MG |
| Ambalaj Miktarı | 14 |
| Antineoplastik ve İmmünomodülatör Ajanlar > İmmünsupresif Ajanlar |
| Yerli ve Beşeri bir ilaçdır. |