TECENTRIQ 840 mg/14 ml inf�zyonluk ��zelti haz�rlamak i�in konsantre K�sa �r�n Bilgisi
{ Atezolizumab }
1. BE�ER� TIBB� �R�N�N ADI
TECENTRIQ 840 mg/14 mL inf�zyonluk ��zelti haz�rlamak i�in konsantre Steril
2. KAL�TAT�F VE KANT�TAT�F B�LE��M
Etkin madde
14 mL konsantre bir flakon i�inde 840 mg atezolizumab i�erir.
Dil�syondan sonra 1 mL sol�syon yakla��k olarak 3,2 mg atezolizumab i�erir (bkz. B�l�m 6.6).
Atezolizumab, rekombinant DNA teknolojisiyle �in hamsteri yumurtal�k h�crelerinde �retilen, bir Fc b�lgesi de�i�tirilmi�, h�manize IgG1 anti-programl� �l�m-ligand� 1 (PD-L1) monoklonal antikorudur.
Yard�mc� maddeler
Yard�mc� maddeler i�in B�l�m 6.1'e bak�n�z.
3. FARMAS�T�K FORMU
�nf�zyonluk ��zelti haz�rlamak i�in steril konsantre Berrak, renksiz ila hafif sar�ms� s�v�
4. KL�N�K �ZELL�KLER
4.1. Terap�tik endikasyonlar
TECENTRIQ'in, t�m�rde PDL1 ekspresyonu %1 ve �zerinde olan (SP142 Ventana �mm�nhistokimya ile), daha �nce kemoterapi almam�� metastatik veya rezeke edilemeyen ��l� negatif meme kanserli (�NMK) hastalar�n tedavisinde nab-paklitaksel ile kombine olarak kullan�m� endikedir. Hastalar �nceden adjuvan kemoterapi alm��larsa hastal�ks�z interval 12 ay ve �zeri olmal�d�r.
4.2. Pozoloji ve uygulama �ekli
TECENTRIQ, kanser tedavisinde deneyimli bir hekimin g�zetimi alt�nda uygulanmal�d�r.
Daha �nce tedavi almam�� �NMK hastalar�, ge�erlili�i g�sterilmi� bir test ile do�rulanan PD-L1 t�m�r ekspresyonuna dayanarak tedavi i�in se�ilmelidir (bkz. B�l�m 5.1).
Pozoloji/uygulama s�kl��� ve s�resi:
�nerilen TECENTRIQ dozu, 100 mg/m nab-paklitakseli takiben intraven�z yoldan uygulanan 840 mg'd�r. Her 28 g�nl�k siklusta, TECENTRIQ 1. ve 15. g�nlerde, nab-paklitaksel ise 1., 8. ve 15. g�nlerde uygulan�r.
Tedavi s�resi:
Klinik faydan�n kaybedilmesine (bkz. B�l�m 5.1) veya y�netilemeyen toksisiteye kadar
hastalar�n TECENTRIQ ile tedavi edilmeleri �nerilmektedir.
Geciken veya atlanan dozlar:
Planlanm�� bir TECENTRIQ dozu atlan�rsa m�mk�n olan en k�sa s�rede uygulanmal�d�r; planlanan sonraki doza kadar beklenmemelidir. Uygulama plan�, dozlar aras�nda uygun bir aral�k korunacak �ekilde ayarlanmal�d�r.
Tedavi s�ras�nda doz modifikasyonlar�:
TECENTRIQ i�in doz azalt�m� �nerilmez.
Doz gecikmesi veya kesilmesi (ayr�ca bkz. B�l�m 4.4 ve 4.8):
Tablo 1: Belirtilen Advers �la� Reaksiyonlar� i�in doz modifikasyon tavsiyesi
Advers reaksiyon | �iddet | Tedavi modifikasyonu |
Pn�moni | 2. derece | TECENTRIQ tedavisine ara verilir.
Olay 12 hafta i�inde 0. derece veya 1. dereceye iyile�ti�inde ve kortikosteroidler g�nde ≤10 mg oral prednizon veya e�de�erine d���r�ld���nde tedaviye devam edilebilir. |
3. veya 4. derece | TECENTRIQ tedavisi tamamen kesilir. | |
Hepatit | 2. derece: (ALT veya AST >3-5 x normalin �st s�n�r� [N�S] veya kan bilirubin >1,5-3 x N�S) | TECENTRIQ tedavisine ara verilir.
Olay 12 hafta i�inde 0. derece veya 1. dereceye iyile�ti�inde ve kortikosteroidler g�nde ≤10 mg oral prednizon veya e�de�erine d���r�ld���nde tedaviye devam edilebilir. |
3. veya 4. derece: (ALT veya AST >5 x N�S veya kan bilirubin >3 x N�S) | TECENTRIQ tedavisi tamamen kesilir. | |
Kolit | 2. veya 3. derece ishal (ba�lang�ca g�re ≥4 d��k�/g�n veya semptomatik kolit) | TECENTRIQ tedavisine ara verilir.
Olay 12 hafta i�inde 0. derece veya 1. dereceye iyile�ti�inde ve kortikosteroidler g�nde ≤10 mg oral prednizon veya e�de�erine d���r�ld���nde tedaviye devam edilebilir. |
4. derece ishal veya kolit (ya�am� tehdit edici; acil m�dahale endike) | TECENTRIQ tedavisi tamamen kesilir. |
Advers reaksiyon | �iddet | Tedavi modifikasyonu |
Hipotiroidizm veya hipertiroidizm | Semptomatik | TECENTRIQ tedavisine ara verilir.
Hipotiroidizm: Semptomlar tiroid replasman tedavisi ile kontrol alt�na al�nd���nda ve TSH d�zeyleri d��meye ba�lad���nda tedaviye devam edilebilir.
Hipertiroidizm: Semptomlar anti-tiroidila�lar� ile kontrol alt�na al�nd���nda ve tiroid fonksiyonu iyile�meye ba�lad���nda tedaviye devam edilebilir. |
Adrenal yetmezlik | Semptomatik | TECENTRIQ tedavisine ara verilir.
Semptomlar 12 hafta i�inde 0. derece veya 1. dereceye iyile�ti�inde ve kortikosteroidler g�nde ≤10 mg oral prednizon veya e�de�erine d���r�ld���nde ve hastan�n durumu replasman tedavisinde stabil hale geldi�inde tedaviye devam edilebilir. |
Hipofizit | 2. veya 3. derece | TECENTRIQ tedavisine ara verilir.
Semptomlar 12 hafta i�inde 0. derece veya 1. dereceye iyile�ti�inde ve kortikosteroidler g�nde ≤10 mg oral prednizon veya e�de�erine d���r�ld���nde ve hastan�n durumu replasman tedavisinde stabil hale geldi�inde tedaviye devam edilebilir. |
4. derece | TECENTRIQ tedavisi tamamen kesilir. | |
Tip 1 diabetes mellitus | 3. veya 4. derece hiperglisemi (a�l�k glikozu >250 mg/dL veya 13,9 mmol/L) | TECENTRIQ tedavisine ara verilir
�ns�lin replasman tedavisinde metabolik kontrol elde |
Advers reaksiyon | �iddet | Tedavi modifikasyonu |
|
| edildi�inde tedaviye devam edilebilir. |
�nf�zyonla ili�kili reaksiyonlar | 1. veya 2. derece | �nf�zyon h�z� azalt�l�r veya kesilir.
Olay d�zeldikten sonra tedavi s�rd�r�lebilir. |
3. veya 4. derece | TECENTRIQ tedavisi tamamen kesilir. | |
D�k�nt� / Ciddi cilt advers reaksiyonlar� | 3. derece
veya ��pheli Stevens-Johnson sendromu (SJS) veya toksik epidermal nekroliz (TEN) | TECENTRIQ tedavisine ara verilir.
Semptomlar 12 hafta i�inde Derece 0 veya Derece 1'e iyile�ti�inde ve kortikosteroidler g�nde ≤ 10 mg prednizon veya e�de�erine d���r�ld���nde tedaviye devam edilebilir. |
| 4. derece
veya do�rulanm�� Stevens- Johnson sendromu (SJS) veya toksik epidermal nekroliz (TEN) | TECENTRIQ tedavisi tamamen kesilir. |
Miyastenik sendrom/ miyastenia gravis, Guillain-Barré sendromu ve meningoensefalit | T�m dereceler | TECENTRIQ tedavisi tamamen kesilir. |
Pankreatit | Serum amilaz veya lipaz d�zeylerinde 3. veya 4. derece y�kselme (> 2 x N�S) veya 2. veya 3. derece pankreatit | TECENTRIQ tedavisine ara verilir.
Serum amilaz ve lipaz d�zeyleri 12 hafta i�inde 0. derece veya 1. dereceye iyile�ti�inde veya pankreatit semptomlar� d�zeldi�inde ve kortikosteroidler g�nde ≤10 mg oral prednizon veya e�de�erine d���r�ld���nde TECENTRIQ ile tedaviye devam edilebilir. |
4. derece veya herhangi bir derecede n�kseden pankreatit | TECENTRIQ tedavisi tamamen kesilir. | |
Miyokardit | 2. derece veya �zeri | TECENTRIQ tedavisi tamamen kesilir. |
Nefrit | 2. derece: (kreatinin seviyesi >1,5 ila 3 x | TECENTRIQ tedavisine ara verilir. |
Advers reaksiyon | �iddet | Tedavi modifikasyonu |
| ba�lang�� veya >1,5 ila 3 x N�S) |
Olay 12 hafta i�inde 0. derece veya 1. dereceye iyile�ti�inde ve kortikosteroidler g�nde ≤10 mg oral prednizon veya e�de�erine d���r�ld���nde tedaviye devam edilebilir. |
3. veya 4. derece: (kreatinin seviyesi >3 x ba�lang�� veya >3 x N�S) | TECENTRIQ tedavisi tamamen kesilir. | |
Miyozit | 2. veya 3. derece | TECENTRIQ tedavisine ara verilir. |
4. derece veya tekrarlayan 3. derece miyozit | TECENTRIQ tedavisi tamamen kesilir. | |
�mm�nite ile ili�kili di�er advers reaksiyonlar | 2. veya 3. derece | Olay 12 hafta i�inde 0. derece veya 1. dereceye iyile�ene ve kortikosteroidler g�nde ≤10 mg oral prednizon veya e�de�erine d���r�lene kadar TECENTRIQ tedavisine ara verilir. |
4. veya 3. derece n�kseden advers olaylar | TECENTRIQ tedavisi tamamen kesilir. (replasman hormonlar�yla kontrol alt�na al�nan endokrinopatiler hari�) |
Not: Toksisite dereceleri, Ulusal Kanser Enstit�s� Advers Olaylar i�in Ortak Terminoloji Kriterleri, Versiyon 4.0'a (NCI-CTCAE v.4) uygundur.
TECENTRIQ ile tedavi edilen hastalara ilac�n riskleri hakk�nda bilgi veren Hasta Uyar� Kartlar� verilmelidir.
Uygulama �ekli:
TECENTRIQ intraven�z kullan�m i�indir. TECENTRIQ inf�zyonlar� intraven�z pu�e veya bolus �eklinde uygulanmamal�d�r.
�lk TECENTRIQ dozu 60 dakika uygulanmal�d�r. �lk inf�zyon iyi tolere edilirse, sonraki t�m inf�zyonlar 30 dakikada uygulanabilir.
T�bbi �r�n�n uygulanmadan �nceden seyreltilmesi ve kullan�m�na ili�kin talimatlar i�in B�l�m
6.6'ya bak�n�z.
�zel pop�lasyonlara ili�kin ek bilgiler:
Karaci�er yetmezli�i:
Pop�lasyon farmakokinetik analizine g�re hafif veya orta derecede karaci�er bozuklu�u olan hastalarda doz ayarlamas� gerekli de�ildir. �iddetlikaraci�er bozuklu�u olan hastalara ili�kin
veri mevcut de�ildir (bkz. B�l�m 5.2).
B�brek yetmezli�i:
Pop�lasyon farmakokinetik analizine g�re hafif veya orta derecede b�brek bozuklu�u olan hastalarda doz ayarlamas� gerekli de�ildir (bkz. B�l�m 5.2). �iddetli b�brek yetmezli�i olan hastalardan elde edilen veriler, bu pop�lasyon hakk�nda sonuca varmak i�in �ok s�n�rl�d�r.
Pediyatrik pop�lasyon:
TECENTRIQ'in 18 ya��ndan k���k �ocuklarda ve adolesanlarda g�venlili�i ve etkilili�i belirlenmemi�tir. Mevcut veriler B�l�m 4.8, 5.1 ve 5.2'de a��klanm��t�r, ancak pozoloji konusunda herhangi bir �neri yap�lamaz.
Geriyatrik pop�lasyon:
Pop�lasyon farmakokinetik analizine g�re ≥ 65 ya��ndaki hastalarda TECENTRIQ doz ayarlamas� gerekli de�ildir (bkz. B�l�m 4.8 ve 5.1).
Do�u ��birli�i Onkoloji Grubu (ECOG) performans durumu skoru ≥ 2:
ECOG performans durumu skoru ≥ 2 olan hastalar �NMK klinik �al��mas�na dahil edilmemi�tir (bkz. B�l�m 4.4 ve 5.1).
4.3. Kontrendikasyonlar
TECENTRIQ'in etkin maddesi atezolizumaba veya B�l�m 6.1'de listelenen yard�mc� maddelerden herhangi birine a��r� duyarl�l��� olan hastalarda kontrendikedir.
4.4. �zel kullan�m uyar�lar� ve �nlemleri
TECENTRIQ ile tedavi s�ras�nda olu�an imm�nite ile ili�kili advers reaksiyonlar�n �o�u ilac�n kesilmesi ve kortikosteroidlerin ve/veya destekleyici tedavinin ba�lat�lmas�yla geri d�nd�r�lebilir olmu�tur. Birden fazla v�cut sistemini etkileyen imm�nite ile ili�kili advers reaksiyonlar g�r�lm��t�r ve bu reaksiyonlar TECENTRIQ'in son dozundan sonra da olu�abilir.
�mm�nite ile ili�kili ��pheli advers reaksiyonlar i�in etiyolojiyi do�rulamak veya di�er nedenleri d��lamak i�in yeterli de�erlendirme yap�lmal�d�r. Advers etkilerin �iddetine ba�l� olarak, TECENTRIQ tedavisine ara verilir ve kortikosteroid uygulan�r. Olay ≤ 1. dereceye iyile�ti�inde kortikosteroid kullan�m� 1 ay boyunca azalt�larak kesilir. �mm�nite ile ili�kili istenmeyen reaksiyonlar�n kortikosteroid kullan�m� ile kontrol edilemedi�i hastalarda, klinik �al��malardan elde edilen s�n�rl� verilere dayanarak, di�er sistemik immunosupresan ajanlar�n kullan�m� d���n�lebilir.
Herhangi bir ≥ 3. derece toksisite ikinci defa ortaya ��karsa ve replasman hormonlar ile kontrol edilen endokrinopatiler hari� herhangi bir 4. derece imm�nite ile ili�kili advers reaksiyon g�r�l�rse TECENTRIQ tedavisi tamamen kesilir (bkz. B�l�m 4.2 ve 4.8).
�mm�nite ile ili�kili pn�moni:
TECENTRIQ ile y�r�t�len klinik �al��malarda �l�mc�l vakalar da dahil olmak �zere pn�moni vakalar� g�zlenmi�tir (bkz. B�l�m 4.8). Hastalar pn�moni belirtileri ve semptomlar� i�in izlenmeli ve imm�n ili�kili pn�monit d���ndaki nedenler hari� b�rak�lmal�d�r.
2. derece pn�moni durumunda TECENTRIQ ile tedaviye ara verilmeli ve g�nde 1-2 mg/kg prednizon veya e�de�eri ile tedavi ba�lat�lmal�d�r. Semptomlar ≤1. dereceye iyile�irse
kortikosteroidler ≥1 ayda azalt�lmal�d�r. Olay 12 hafta i�inde ≤1. dereceye iyile�irse ve kortikosteroidler g�nde ≤10 mg oral prednizon veya e�de�erine d���r�l�rse TECENTRIQ ile tedaviye devam edilebilir. 3. veya 4. derece pn�moni durumunda TECENTRIQ ile tedavi kal�c� olarak b�rak�lmal�d�r.
�mm�nite ile ili�kili hepatit:
TECENTRIQ ile y�r�t�len klinik �al��malarda baz�lar� �l�mc�l sonu�lara yol a�an hepatit vakalar� g�zlenmi�tir (bkz. B�l�m 4.8). Hastalar hepatit belirtileri ve semptomlar� i�in izlenmelidir.
Aspartat aminotransferaz (AST), alanin aminotransferaz (ALT) ve bilirubin, TECENTRIQ ile tedaviye ba�lamadan �nce ve tedavi s�ras�nda periyodik olarak ve klinik de�erlendirmeye g�re endike oldu�u gibi izlenmelidir.
2. derece anormallik (ALT veya AST >3-5 x N�S veya kan bilirubin >1,5-3 x N�S) 5-7 g�nden uzun s�re devam ederse TECENTRIQ ile tedaviye ara verilmeli ve g�nde 1-2 mg/kg prednizon veya e�de�eri ile tedavi ba�lat�lmal�d�r. Olaylar ≤1. dereceye iyile�irse kortikosteroidler ≥1 ayda azalt�lmal�d�r.
Olay 12 hafta i�inde ≤1. dereceye iyile�irse ve kortikosteroidler g�nde ≤10 mg oral prednizon veya e�de�erine d���r�l�rse TECENTRIQ ile tedaviye devam edilebilir. 3. veya 4. derece olaylarda (ALT veya AST >5 x N�S veya kan bilirubin >3 x N�S) TECENTRIQ ile tedavi kal�c� olarak b�rak�lmal�d�r.
�mm�nite ile ili�kili kolit:
TECENTRIQ ile y�r�t�len klinik �al��malarda ishal veya kolit vakalar� g�zlenmi�tir (bkz. B�l�m 4.8). Hastalar kolit belirtileri ve semptomlar� i�in izlenmelidir.
2. veya 3. derece ishal (ba�lang�ca g�re ≥4 d��k�/g�n art��) veya kolit (semptomatik) durumunda TECENTRIQ ile tedaviye ara verilmelidir. 2. derece ishal veya kolit durumunda semptomlar
>5 g�n devam ederse veya n�ksederse, g�nde 1-2 mg/kg prednizon veya e�de�eri ile tedavi ba�lat�lmal�d�r. 3. derece ishal veya kolit durumunda intraven�z kortikosteroidlerle (1-2 mg/kg/g�n metilprednizolon veya e�de�eri) tedavi ba�lat�lmal� ve iyile�me sonras�nda oral kortikosteroidlere (g�nde 1-2 mg/kg prednizon veya e�de�eri) ge�ilmelidir. Semptomlar ≤1. dereceye iyile�irse kortikosteroidler ≥1 ayda azalt�lmal�d�r. Olay 12 hafta i�inde ≤1. dereceye iyile�irse ve kortikosteroidler g�nde ≤10 mg oral prednizon veya e�de�erine d���r�l�rse TECENTRIQ ile tedaviye devam edilebilir. 4. derece (ya�am� tehdit edici; acil m�dahale endike) ishal veya kolit durumunda TECENTRIQ ile tedavi kal�c� olarak b�rak�lmal�d�r.
�mm�nite ile ili�kili endokrinopatiler:
TECENTRIQ ile y�r�t�len klinik �al��malarda hipotiroidizm, hipertiroidizm, adrenal yetmezlik, hipofizit ve diyabetik ketoasidoz dahil olmak �zere tip 1 diabetes mellitus vakalar� g�zlenmi�tir (bkz. B�l�m 4.8).
Hastalar endokrinopatilerin klinik belirtileri ve semptomlar� i�in izlenmelidir. Tiroid fonksiyonu TECENTRIQ ile tedavi �ncesinde ve tedavi s�ras�nda periyodik olarak izlenmelidir. Ba�lang��ta anormal tiroid fonksiyon testleri olan hastalar�n uygun �ekilde tedavi edilmesi d���n�lmelidir.
Anormal tiroid fonksiyonu olan asemptomatik hastalar TECENTRIQ alabilir. Semptomatik
hipotiroidizm durumunda TECENTRIQ ile tedaviye ara verilmeli ve tiroid hormonu replasman� gerekti�inde ba�lat�lmal�d�r. �zole hipotiroidizm kortikosteroidler kullan�lmadan replasman tedavisi ile y�netilebilir. Semptomatik hipertiroidizm durumunda TECENTRIQ ile tedaviye ara verilmeli ve bir anti-tiroid t�bbi �r�n gerekti�i gibi ba�lat�lmal�d�r. Semptomlar kontrol alt�na al�nd���nda ve tiroid fonksiyonu iyile�ti�inde TECENTRIQ ile tedaviye devam edilebilir.
Semptomatik adrenal yetmezlik durumunda TECENTRIQ ile tedaviye ara verilmeli ve intraven�z kortikosteroid (g�nde 1-2 mg/kg metilprednizolon veya e�de�eri) ile tedavi ba�lat�lmal�d�r. Semptomlar iyile�ti�inde g�nde 1-2 mg/kg oral prednizon veya e�de�eri ile tedavi uygulanmal�d�r. Semptomlar ≤1. dereceye iyile�irse kortikosteroidler ≥1 ayda azalt�lmal�d�r. Olay 12 hafta i�inde ≤1. dereceye iyile�irse ve kortikosteroidler g�nde ≤10 mg oral prednizon veya e�de�erine d���r�l�rse ve hastan�n durumu replasman tedavisinde stabilse (gerektiyse) tedaviye devam edilebilir.
2. veya 3. derece hipofizit i�in TECENTRIQ ile tedaviye ara verilmeli ve intraven�z kortikosteroidler (1 ila 2 mg/kg/g�n metilprednizolon veya e�de�eri) ile tedavi ba�lat�lmal� ve ihtiyaca g�re hormon replasman tedavisi ba�lat�lmal�d�r. Belirtiler d�zeldi�inde 1-2 mg/kg/g�n prednizon veya e�de�eri ile tedavi uygulanmal�d�r. Semptomlar ≤1. dereceye kadar y�kselirse, kortikosteroidler ≥ 1 ay boyunca azalt�larak kesilebilir. Olay, 12 hafta i�inde ≤1. dereceye y�kselir ve kortikosteroidler g�nde ≤ 10 mg prednizona veya e�de�erine d���r�l�rse ve hasta yedek tedavide (e�er gerekliyse) stabil kal�rsa, tedaviye devam edilebilir. 4. derece hipofizit i�in TECENTRIQ tedavisine ara verilmelidir.
Tip 1 diabetes mellitus i�in ins�lin tedavisi ba�lat�lmal�d�r. ≥3. derece hiperglisemi (a�l�k glikozu >250 mg/dL veya 13,9 mmol/L) durumunda TECENTRIQ ile tedaviye ara verilmelidir. �ns�lin replasman tedavisinde metabolik kontrol elde edilirse TECENTRIQ ile tedaviye devam edilebilir.
�mm�nite ile ili�kili meningoensefalit:
TECENTRIQ ile y�r�t�len klinik �al��malarda meningoensefalit g�zlenmi�tir (bkz. B�l�m 4.8). Hastalar menenjit veya ensefalitin klinik belirtileri ve semptomlar� i�in izlenmelidir.
Herhangi bir derece menenjit veya ensefalit durumunda TECENTRIQ ile tedavi kal�c� olarak b�rak�lmal�d�r. �ntraven�z kortikosteroidler (g�nde 1-2 mg/kg metilprednizolon veya e�de�eri) ile tedavi ba�lat�lmal� ve hastan�n durumu iyile�ti�inde 1-2 mg/kg oral prednizon veya e�de�erine ge�ilmelidir.
�mm�nite ile ili�kili n�ropatiler:
TECENTRIQ tedavisi g�ren hastalarda ya�am� tehdit edici olabilen miyastenik sendrom/miyastenia gravis veya Guillain-Barré sendromu g�zlenmi�tir. Hastalar motor ve duyusal n�ropati semptomlar� i�in izlenmelidir.
Herhangi bir derece miyastenik sendrom/miyastenia gravis veya Guillain-Barré sendromu durumunda TECENTRIQ ile tedavi kal�c� olarak b�rak�lmal�d�r. G�nde 1-2 mg/kg dozda oral prednizon veya e�de�eriyle sistemik kortikosteroidlerin ba�lat�lmas� d���n�lmelidir.
�mm�nite ile ili�kili pankreatit:
TECENTRIQ ile y�r�t�len klinik �al��malarda serum amilaz ve lipaz d�zeylerinde art��lar dahil olmak �zere pankreatit g�zlenmi�tir (bkz. B�l�m 4.8). Hastalar akut pankreatiti
d���nd�ren belirtiler ve semptomlar i�in yak�ndan izlenmelidir.
Serum amilaz veya lipaz d�zeylerinde ≥3. derece art�� (>2 x N�S) veya 2. veya 3. derece pankreatit durumunda TECENTRIQ ile tedaviye ara verilmeli ve intraven�z kortikosteroidler (g�nde 1-2 mg/kg metilprednizolon veya e�de�eri) ile tedavi ba�lat�lmal�d�r. Semptomlar iyile�ti�inde g�nde 1-2 mg/kg oral prednizon veya e�de�eri ile tedavi uygulanmal�d�r. Serum amilaz ve lipaz d�zeyleri 12 hafta i�inde ≤1. dereceye iyile�ti�inde veya pankreatit semptomlar� d�zeldi�inde ve kortikosteroidler g�nde ≤10 mg oral prednizon veya e�de�erine d���r�ld���nde TECENTRIQ ile tedaviye devam edilebilir. 4. derece veya herhangi bir derecede n�kseden pankreatit durumunda TECENTRIQ ile tedavi kal�c� olarak b�rak�lmal�d�r.
�mm�nite ile ili�kili miyokardit:
TECENTRIQ ile y�r�t�len klinik �al��malarda �l�mc�l vakalar da dahil olmak �zere miyokardit g�zlemlenmi�tir (bkz. B�l�m 4.8). Hastalar miyokarditi d���nd�ren belirtiler ve semptomlar i�in yak�ndan izlenmelidir. Miyokardit ayr�ca miyozitin klinik bir belirtisi olabilir ve buna uygun olarak tedavi edilmelidir.
Kardiyak veya kardiyopulmoner semptomlar� olan hastalar, uygun �nlemlerin erken evrede ba�lat�labilmesi i�in potansiyel miyokardit a��s�ndan de�erlendirilmelidir. Miyokardit ��phesi varsa, TECENTRIQ tedavisine ara verilmeli, g�nde 1 ila 2 mg/kg v�cut a��rl���/g�n prednizon veya e�de�eriyle sistemik kortikosteroidler ba�lanmal� ve g�ncel klinik k�lavuzlara g�re tan�sal �al��mayla birlikte derhal kardiyoloji kons�ltasyonu yap�lmal�d�r. Miyokardit tan�s� konuldu�unda, derece ≥ 2 miyokardit i�in atezolizumab tedavisi kal�c� olarak kesilmelidir (bkz. B�l�m 4.2).
�mm�nite ile ili�kili nefrit:
TECENTRIQ ile yap�lan klinik �al��malarda nefrit g�zlenmi�tir (bkz. B�l�m 4.8). Hastalar b�brek fonksiyonundaki de�i�iklikler a��s�ndan izlenmelidir.
2. derece nefrit durumunda TECENTRIQ tedavisine ara verilmeli ve g�nde 1 ila 2 mg/kg v�cut a��rl���/g�n prednizon veya e�de�eriyle sistemik kortikosteroidler ba�lanmal�d�r. Olay 12 hafta i�inde derece ≤ 1'e y�kselirse ve kortikosteroidler g�nde ≤ 10 mg prednizon veya e�de�erine d���r�l�rse atezolizumab ile tedaviye devam edilebilir. 3. veya 4. derece nefrit i�in TECENTRIQ ile tedavi kal�c� olarak b�rak�lmal�d�r.
�mm�nite ile ili�kili miyozit:
Atezolizumab ile �l�mc�l vakalar dahil miyozit vakalar� g�zlenmi�tir (bkz. B�l�m 4.8). Hastalar miyozit belirti ve semptomlar� a��s�ndan izlenmelidir. Olas� miyoziti olan hastalar miyokardit belirtileri a��s�ndan izlenmelidir.
Bir hastada miyozit belirti ve semptomlar� geli�irse, yak�ndan takip edilmeli ve hasta zaman kaybetmeden de�erlendirme ve tedavi i�in bir uzmana y�nlendirilmelidir. 2. veya 3. derece miyozit durumunda TECENTRIQ tedavisine ara verilmeli ve kortikosteroid tedavisi (1-2 mg/kg v�cut a��rl���/g�n prednizon veya e�de�eri) ba�lat�lmal�d�r. Semptomlar derece ≤ 1'e y�kselirse, klinik olarak belirtildi�i gibi kortikosteroidler azalt�lmal�d�r. Olay 12 hafta i�inde derece ≤ 1'e y�kselirse ve kortikosteroidler g�nde ≤ 10 mg oral prednizon veya e�de�erine d���r�l�rse TECENTRIQ ile tedaviye devam edilebilir. 4. veya 3. derece tekrarlayan miyozit durumunda veya ba�lang��tan sonraki 12 hafta i�inde kortikosteroid dozu g�nde ≤ 10 mg prednizon e�de�erine d���r�lemedi�inde TECENTRIQ ile tedavi kal�c� olarak kesilmelidir.
�mm�nite ile ili�kili �iddetli kutan�z advers reaksiyonlar:
TECENTRIQ ile tedavi edilen hastalarda Stevens-Johnson sendromu (SJS) ve toksik epidermal nekroliz (TEN) vakalar� dahil olmak �zere imm�nite ile ili�kili �iddetli kutan�z advers reaksiyonlar (SCAR'lar) bildirilmi�tir. Hastalar, ��pheli �iddetli cilt reaksiyonlar a��s�ndan izlenmeli ve di�er sebepler d��lanmal�d�r. ��pheli SCAR'lar varl���nda hastalar ileri tan� ve tedavi i�in bir uzmana y�nlendirilmelidir.
Advers reaksiyonun derecesine ba�l� olarak, 3. derece cilt reaksiyonu durumunda TECENTRIQ ile tedaviye ara verilmeli ve g�nde 1-2 mg/kg prednizon veya e�de�eri ile sistemik kortikosteroidler ba�lanmal�d�r. Olay 12 hafta i�inde derece ≤ 1'e iyile�ir ve kortikosteroidler g�nde ≤ 10 mg prednizon veya e�de�erine d���r�l�rse TECENTRIQ ile tedaviye devam edilebilir. 4. derece cilt reaksiyonlar� i�in TECENTRIQ tedavisi kal�c� olarak b�rak�lmal� ve kortikosteroidler uygulanmal�d�r.
SJS veya TEN ��phesi olan hastalarda TECENTRIQ tedavisine ara verilmelidir. Do�rulanm�� SJS veya TEN durumunda TECENTRIQ tedavisi kal�c� olarak kesilmelidir.
Daha �nce di�er imm�n sistemi uyar�c� antikanser ajanlarla tedavi s�ras�nda ciddi veya ya�am� tehdit eden bir kutan�z advers reaksiyon deneyimleyen bir hastada TECENTRIQ kullan�m� d���n�l�rken dikkatli olunmal�d�r.
�mm�nite ile ili�kili di�er advers reaksiyonlar:
TECENTRIQ'in etki mekanizmas� g�z �n�ne al�nd���nda, enfektif olmayan sistit dahil olmak �zere ba����kl�kla ilgili di�er potansiyel advers reaksiyonlar meydana gelebilir.
Di�er nedenleri d��lamak i�in ba����kl�kla ilgili t�m ��pheli advers reaksiyonlar de�erlendirilmelidir. Hastalar, ba����kl�kla ilgili advers reaksiyonlar�n belirti ve semptomlar� a��s�ndan izlenmeli ve reaksiyonun ciddiyetine ba�l� olarak, klinik olarak belirtildi�i gibi tedavi de�i�iklikleri ve kortikosteroidlerle g�zetim alt�nda tutulmal�d�r (bkz. B�l�m 4.2 ve B�l�m 4.8).
�nf�zyon ile ili�kili reaksiyonlar:
TECENTRIQ ile y�r�t�len klinik �al��malarda inf�zyon ile ilgili reaksiyonlar g�zlenmi�tir (bkz. B�l�m 4.8).
1. veya 2. derece inf�zyon ile ili�kili reaksiyon g�r�ld���nde inf�zyon h�z� d���r�lmeli veya tedaviye ara verilmelidir. 3. veya 4. derece inf�zyon ile ilgili reaksiyon g�r�ld���nde TECENTRIQ tedavisi tamamen sonland�r�lmal�d�r. 1. veya 2. derece inf�zyon ile ili�kili reaksiyon g�r�len hastalar yak�ndan izlenerek TECENTRIQ almaya devam edebilir; bu hastalarda antipiretik ve antihistaminiklerle premedikasyon de�erlendirilebilir.
Hastal�k spesifik �nlemler:
Metastatik �NMK'de TECENTRIQ'in nab-paklitaksel ile kombinasyon halinde kullan�m�
TECENTRIQ ve nab-paklitaksel ile tedavi s�ras�nda ortaya ��kan n�tropeni ve periferik n�ropatiler, nab-paklitakselin kesilmesiyle geri d�n���ml� olabilir. Doktorlar, bu ilac�n �zel �nlemleri ve kontrendikasyonlar� i�in nab-paklitaksel K�sa �r�n Bilgisine (K�B) ba�vurmal�d�r.
Klinik �al��malardan d��lanan hastalar:
Otoimm�n hastal�k ge�mi�i, pn�moni ge�mi�i, aktif beyin metastaz�, HIV, hepatit B veya hepatit C enfeksiyonu, �nemli kardiyovask�ler hastal�k ve yetersiz hematolojik ve u� organ i�levi olan hastalar TECENTRIQ ile y�r�t�len klinik �al��malara al�nmam��t�r. Kay�ttan �nceki 28 g�n i�inde canl�, aten�e a�� uygulanan hastalar; 4 hafta i�inde sistemik imm�n sistemi uyar�c� ajan alan hastalar veya �al��maya giri�ten 2 hafta �nce sistemik immunosupresif t�bbi �r�n kullanan hastalar; �al��ma tedavisinin ba�lamas�ndan �nceki 2 hafta i�inde terap�tik oral veya intraven�z antibiyotik kullanan hastalar klinik �al��malara al�nmam��t�r.
Hasta Uyar� Kart�:
TECENTRIQ re�ete eden hekimin hasta ile TECENTRIQ tedavisinin risklerini konu�mas� gerekmektedir. TECENTRIQ ile tedavi edilen hastalara ilac�n riskleri hakk�nda bilgi veren Hasta Uyar� Kartlar� verilmeli ve kart� her zaman yanlar�nda ta��malar� s�ylenmelidir.
Biyoteknolojik �r�nlerin takip edilebilirli�inin sa�lanmas� i�in uygulanan �r�n�n ticari ismi ve seri numaras� mutlaka hasta dosyas�na kaydedilmelidir.
4.5. Di�er t�bbi �r�nler ile etkile�imler ve di�er etkile�im �ekilleri
TECENTRIQ ile herhangi bir resmi farmakokinetik ila� etkile�imi �al��mas� yap�lmam��t�r. TECENTRIQ dola��mdan katabolizma ile temizlendi�i i�in metabolik ila�-ila� etkile�imleri beklenmemektedir.
TECENTRIQ ile tedaviye ba�lamadan �nce, TECENTRIQ'in farmakodinamik aktivitesine ve etkilili�ine yapabilecekleri potansiyel etkiler nedeniyle sistemik kortikosteroidlerin veya immunosupresanlar�n kullan�lmas�ndan ka��n�lmal�d�r. Bununla birlikte, sistemik kortikosteroidler veya di�er immunosupresif maddeler, TECENTRIQ tedavisine ba�lad�ktan sonra imm�nite ile ili�kili advers reaksiyonlar�n tedavisinde kullan�labilir (bkz. B�l�m 4.4).
�zel pop�lasyonlara ili�kin ek bilgiler:
TECENTRIQ ile herhangi bir farmakokinetik ila� etkile�imi �al��mas� yap�lmam��t�r.
Pediyatrik pop�lasyon:
TECENTRIQ ile pediyatrik pop�lasyonda herhangi bir farmakokinetik ila� etkile�imi �al��mas� yap�lmam��t�r.
4.6. Gebelik ve laktasyon
:Gebelik kategorisi: D
�ocuk do�urma potansiyeli bulunan kad�nlar/do�um kontrol� (kontrasepsiyon):
�ocuk do�urma potansiyeline sahip kad�nlar TECENTRIQ ile tedavi s�ras�nda ve tedaviden 5 ay sonraya kadar etkili bir do�um kontrol y�ntemi kullanmal�d�r.
Gebelik d�nemi:
Atezolizumab�n fet�s �zerinde zararl� farmakolojik etkileri bulunmaktad�r. Atezolizumab ile geli�imsel �al��malar ve �reme �al��malar� yap�lmam��t�r. Hayvan �al��malar�yla, PD-L1/PD-1 yolak inhibisyonunun fare veya s��an gebelik modellerinde imm�nite ile ili�kili, fet�s �l�m�yle sonu�lanan fet�s geli�iminin reddine sebep oldu�u g�sterilmi�tir (bkz. B�l�m 5.3). �nsanlara
y�nelik potansiyel risk bilinmemektedir ancak hayvan �al��malar�ndan al�nan sonu�lar, etki mekanizmas�na ba�l� olarak, gebelik d�neminde atezolizumab uygulamas�n�n artm�� d���k ve �l� do�um oranlar� dahil olmak �zere f�tal zarara sebep olabilece�ini g�stermektedir.
Atezolizumab bir insan G1 imm�noglob�linidir (IgG1) ve IgG1'in plasenta bariyerini a�t��� bilinmektedir. Bu nedenle, atezolizumab�n anneden geli�mekte olan fet�se ge�me potansiyeli bulunmaktad�r.
Gebe kad�nlar�n klinik durumu atezolizumab ile tedavi gerektirmedik�e gebelik s�ras�nda TECENTRIQ kullan�lmamal�d�r.
Laktasyon d�nemi:
Atezolizumab�n anne s�t�ne ge�ip ge�medi�i bilinmemektedir. Atezolizumab bir monoklonal antikordur ve ilk gelen s�tte bulunmas� ve daha sonra da az miktarda s�tte bulunmas� beklenmektedir. Yeni do�anlar ve infantlar �zerindeki risk d��lanamaz. Emzirmenin �ocuk i�in faydalar� ve tedavinin anne i�in faydalar� dikkate al�narak emzirmenin kesilmesi veya TECENTRIQ tedavisinin kesilmesi kararla�t�r�lmal�d�r.
�reme yetene�i/fertilite:
Atezolizumab�n fertilite �zerindeki olas� etkilerine ili�kin veri bulunmamaktad�r. Atezolizumab�n do�urganl�k �zerindeki etkisini de�erlendirme ama�l� reprod�ktif ve geli�imsel toksisite �al��malar� yap�lmam��t�r. Bununla birlikte, 26 haftal�k tekrarlanan doz toksisitesi �al��mas�na dayal� olarak, atezolizumab�n, �nerilen dozu alan hastalarda e�ri alt� alan� (EAA)'n�n yakla��k 6 kat� tahmini bir EAA'da adet d�ng�leri �zerinde bir etkiye sahip oldu�u ve bu etkinin geri d�n���ml� oldu�u g�r�lm��t�r (bkz. B�l�m 5.3). Erkek �reme organlar� �zerinde etki g�r�lmemi�tir.
4.7. Ara� ve makine kullan�m� �zerindeki etkiler
TECENTRIQ'in ara� ve makine kullanma yetene�i �zerinde d���k d�zeyde etkisi vard�r. Yorgunluk hisseden hastalara semptomlar hafifleyene kadar ara� ve makine kullanmamalar� tavsiyesinde bulunulmal�d�r (bkz. B�l�m 4.8).
4.8. �stenmeyen etkiler
G�venlilik profilinin �zeti:
TECENTRIQ monoterapisinin g�venlili�i, birden fazla t�m�r tipinde 4349 hastadan toplanan verilere dayanmaktad�r. En yayg�n advers reaksiyonlar, (>%10) yorgunluk (%30,1), i�tah azalmas� (%21,3), bulant� (%20), d�k�nt� (%19,3), ate� (%19,0), �ks�r�k (%18,6), ishal (%18),
dispne (%17,2), artralji (%16,7), asteni (%13,2), ka��nt� (%13,2), s�rt a�r�s� (%12,8), kusma (%12,5), idrar yolu enfeksiyonu (%11,5) ve ba� a�r�s� (%10,3) olmu�tur. TECENTRIQ monoterapi �al��malar�n�n tan�m� i�in, TECENTRIQ 1200 mg/20 mL inf�zyonluk ��zelti haz�rlamak i�in konsantre'nin K�sa �r�n Bilgisi'ne bak�n�z.
Di�er t�bbi �r�nlerle kombinasyon halinde verilen TECENTRIQ'in g�venlili�i, birden fazla t�m�r tipinde 4535 hastada de�erlendirilmi�tir. En yayg�n advers reaksiyonlar (≥ %20) anemi (%36,8), n�tropeni (%36,6), bulant� (%35,5), yorgunluk (%33,1), alopesi (%28,1), d�k�nt�
(%27,8), ishal (27,6), trombositopeni (%27,1), kab�zl�k (%25,8), i�tah azalmas� (%24,7) ve periferik n�ropati (%24,4)dir.
Ciddi advers reaksiyonlarla ilgili daha fazla bilgi i�in bkz. B�l�m 4.4.
Advers reaksiyonlar�n tablo halinde listesi
Advers �la� Reaksiyonlar� (ADR), TECENTRIQ monoterapisi ve kombinasyon terapisi i�in Tablo 2'de MedDRA sistem organ s�n�f�na (SOC) ve s�kl�k kategorilerine g�re a�a��da listelenmi�tir.
A�a��daki s�kl�k kategorileri kullan�lm��t�r:
�ok yayg�n (≥1/10), yayg�n (≥1/100 ila <1/10), yayg�n olmayan (≥1/1000 ila <1/100), seyrek (≥1/10.000 ila <1/1000), �ok seyrek (<1/10.000), bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor). Her bir s�kl�k grubunda advers reaksiyonlar azalan ciddiyet s�ras�na g�re verilmektedir.
Tablo 2: TECENTRIQ ile tedavi edilen hastalarda meydana gelen advers reaksiyonlar�n �zeti
| TECENTRIQ monoterapisi | TECENTRIQ kombinasyon tedavisi |
Enfeksiyonlar ve enfestasyonlar | ||
�ok yayg�n | �drar yollar� enfeksiyonu | Akci�er enfeksiyonu |
Yayg�n |
| Sepsis |
Kan ve lenf listemi hastal�klar� | ||
�ok yayg�n |
| Anemi, trombositopeni, n�tropeni, l�kopeni |
Yayg�n | Trombositopeni | Lenfopeni |
Ba����kl�k sistemi hastal�klar� | ||
Yayg�n | �nf�zyonla ili�kili reaksiyon | �nf�zyonla ili�kili reaksiyon |
Endokrin hastal�klar | ||
�ok yayg�n |
| Hipotiroidizm |
Yayg�n | Hipotiroidizm, hipertiroidizm | Hipertiroidizm |
Yayg�n olmayan | Diabetes mellitus, adrenal yetmezlik |
|
Seyrek | Hipofizit |
|
Metabolizma ve beslenme hastal�klar� | ||
�ok yayg�n | ��tah kayb� | ��tah kayb� |
Yayg�n | Hipokalemi, hiponatremi, hiperglisemi | Hipokalemi, hiponatremi, hipomagnezemi |
Sinir sistemi hastal�klar� | ||
�ok yayg�n | Ba� a�r�s� | Periferal n�ropati, ba�a�r�s� |
Yayg�n |
| Bay�lma, ba� d�nmesi |
Yayg�n olmayan | Guillain-Barré sendromu, meningoensefalit |
|
Seyrek | Miyastenik sendrom |
|
G�z hastal�klar� | ||
Seyrek | �veit |
|
Kardiyak hastal�klar | ||
Seyrek | Miyokardit |
|
Vask�ler hastal�klar | ||
�ok yayg�n |
| Hipertansiyon |
Yayg�n | Hipotansiyon |
|
Solunum, g���s bozukluklar� ve mediastinal hastal�klar | ||
�ok yayg�n | Dispne, �ks�r�k | Dispne, �ks�r�k, nazofarenjit |
Yayg�n | Pn�moni, hipoksi, nazofarenjit | Disfoni |
Gastrointestinal hastal�klar | ||
�ok yayg�n | Mide bulant�s�, kusma, ishal | Mide bulant�s�, kusma, ishal, kab�zl�k |
Yayg�n | Kolit, kar�n a�r�s�, disfaji, a��z ve yutak a�r�s� | Stomatit, disguzi |
Yayg�n olmayan | Pankreatit |
|
Hepatobiliyer hastal�klar� | ||
Yayg�n | AST art���, ALT art���, hepatit | AST art���, ALT art��� |
Deri ve deri alt� doku hastal�klar� | ||
�ok yayg�n | D�k�nt�, pr�rit | D�k�nt�, pr�rit, alopesi |
Yayg�n | Kuru cilt |
|
Yayg�n olmayan | Ciddi cilt advers reaksiyonlar�, sedef hastal��� | Ciddi cilt advers reaksiyonlar�, sedef hastal��� |
Seyrek | Pemfigoid | Pemfigoid |
Kas-iskelet bozukluklar�, ba� doku ve kemik hastal�klar� | ||
�ok yayg�n | Artralji, s�rt a�r�s� | Artralji, kas-iskelet a�r�s�, s�rt a�r�s� |
Yayg�n | Kas-iskelet a�r�s� |
|
Yayg�n olmayan | Miyozit |
|
B�brek ve idrar yolu hastal�klar� | ||
Yayg�n | Kan kreatininin artmas� | Protein�ri, kan kreatininin artmas� |
Yayg�n olmayan | Nefrit |
|
Bilinmiyor | Bula��c� olmayan sistit |
|
Genel bozukluklar ve uygulama b�lgesine ili�kin hastal�klar | ||
�ok yayg�n | Ate�, yorgunluk, asteni | Ate�, yorgunluk, asteni, periferik �dem |
Yayg�n | Grip benzeri hastal�k, titreme |
|
Ara�t�rmalar | ||
Yayg�n |
| Kan alkalin fosfataz art��� |
anormal, tri-iyodotironin serbest azalm��, tri-iyodotironin serbest artm��, sessiz tiroidit, kronik tiroidit raporlar�n� i�erir.
Hipoksi, oksijen sat�rasyonunda azalma, pOazalmas� raporlar�n� i�erir.
Se�ilen advers reaksiyonlar�n a��klamas�:
A�a��daki veriler, klinik a��dan anlaml� advers reaksiyonlarla ilgili olarak TECENTRIQ monoterapisine maruziyeti yans�t�r (bkz. B�l�m 5.1). Kombinasyon tedavisi olarak verildi�inde TECENTRIQ i�in se�ilen advers reaksiyonlarla ilgili ayr�nt�lar, yaln�zca
TECENTRIQ monoterapisine k�yasla klinik a��dan anlaml� farkl�l�klar�n bildirilmesi durumunda sunulmaktad�r. Bu advers reaksiyonlar i�in y�netim k�lavuzlar� B�l�m 4.2 ve 4.4'te tan�mlanm��t�r.
�mm�nite ile ili�kili pn�moni:
Pn�moni, TECENTRIQ monoterapisi g�ren hastalar�n %3'�nde (130/4.349) meydana gelmi�tir. 130 hasta i�inde iki �l�mc�l olay olmu�tur. Ba�lang�ca kadar ge�en medyan s�re 4 ay (aral�k: 3 g�n – 29,8 ay) olmu�tur. Medyan s�re 1,6 ay (aral�k: 1 g�n – 27,8+ ay; "+" sans�rlenmi� bir de�eri g�sterir) olmu�tur. Pn�moni, 29 hastada (%0,7) TECENTR�Q'in b�rak�lmas�na yol a�m��t�r. Kortikosteroid kullan�m� gerektiren pn�moni, TECENTRIQ monoterapisi g�ren hastalar�n %1,7'sinde (76/4.349) meydana gelmi�tir.
�mm�nite ile ili�kili hepatit:
Hepatit, TECENTRIQ monoterapisi g�ren hastalar�n %1,7'sinde (75/4.349) meydana gelmi�tir. 75 hasta i�inde iki �l�mc�l olay olmu�tur. Ba�lang�ca kadar ge�en medyan s�re 1,6 ay (aral�k: 7 g�n – 18,8 ay) olmu�tur. Medyan s�re 2,1 ay (aral�k: 1 g�n – 22+ ay; "+" sans�rlenmi� bir de�eri g�sterir) olmu�tur. Hepatit, 13 hastada (%0,3) atezolizumab�n b�rak�lmas�na yol a�m��t�r. Kortikosteroid kullan�m� gerektiren hepatit, TECENTRIQ monoterapisi g�ren hastalar�n %0,5'inde (22/4.349) meydana gelmi�tir.
�mm�nite ile ili�kili kolit:
Kolit, TECENTRIQ monoterapisi g�ren hastalar�n %1,1'inde (50/4.349) meydana gelmi�tir. Ba�lang�ca kadar ge�en medyan s�re 5,1 ay (aral�k: 15 g�n – 17,2 ay) olmu�tur. Medyan s�re 1,2 ay (aral�k: 1 g�n – 35,9+ ay; "+" sans�rlenmi� bir de�eri g�sterir) olmu�tur. Kolit, 17 hastada (%0,4) atezolizumab�n b�rak�lmas�na yol a�m��t�r. Kortikosteroid kullan�m� gerektiren kolit TECENTRIQ monoterapisi g�ren hastalar�n %0,6's�nda (24/4.349) meydana gelmi�tir.
�mm�nite ile ili�kili endokrinopatiler:
Tiroid bozukluklar�
Hipotiroidizm, TECENTRIQ monoterapisi g�ren hastalar�n % 7,6's�nda (331/4.349) meydana gelmi�tir. Ba�lang�ca kadar ge�en medyan s�re 4,3 ay (aral�k: 1 g�n – 34,5 ay) olmu�tur.
Hipertiroidizm, TECENTRIQ monoterapisi g�ren hastalar�n %2,1'inde (93/4.349) meydana gelmi�tir. Ba�lang�ca kadar ge�en medyan s�re 2,6 ay (aral�k: 1 g�n – 24,3 ay) olmu�tur.
Adrenal yetmezlik
Adrenal yetmezlik, TECENTRIQ monoterapisi g�ren hastalar�n %0,5'inde (21/4.349) meydana gelmi�tir. Ba�lang�ca kadar ge�en medyan s�re 6,1 ay (aral�k: 2 g�n – 21,4 ay) olmu�tur. Adrenal yetmezlik nedeniyle 5 (%0,1) hastada TECENTR�Q kullan�m� b�rak�lm��t�r. Kortikosteroid kullan�m� gerektiren adrenal yetmezlik TECENTRIQ monoterapisi g�ren hastalar�n %0,4'�nde (17/4.349) meydana gelmi�tir.
Hipofizit
Hipofizit, TECENTRIQ monoterapisi g�ren hastalar�n %0,1'inden az�nda (4/4.349) meydana gelmi�tir. Ba�lang�ca kadar ge�en medyan s�re 6,1 ay (aral�k: 23 g�n - 13,7 ay'd�r) olmu�tur. �� hastada (<%0,1) kortikosteroid kullan�m� gerekmi�tir ve 1 (<%0,1) hastada TECENTRIQ tedavisi durdurulmu�tur.
Hipofizit, TECENTRIQ ile kombinasyon halinde nab-paklitaksel ve karboplatin tedavisi g�ren hastalar�n %0,4'�nde (2/473) meydana gelmi�tir. Ba�lang�ca kadar ge�en medyan s�re 5,2 ay (aral�k: 5,1 - 5,3 ay) olmu�tur. Her iki hastada da kortikosteroid kullan�m� gerekmi�tir.
Diabetes mellitus
Diabetes mellitus, TECENTRIQ monoterapisi g�ren hastalar�n % 0,5'inde (20/4.349) meydana gelmi�tir. Medyan s�re 5,5 ay (aral�k: 4 g�n – 29 ay) olmu�tur. Diabetes mellitus, <%0,1 (3/4.349) hastada TECENTRIQ'in b�rak�lmas�na yol a�m��t�r.
�mm�nite ile ili�kili meningoensefalit:
Menenjit, TECENTRIQ monoterapisi g�ren hastalar�n %0,4'�nde (18/4.349) meydana gelmi�tir. Ba�lang�ca kadar ge�en s�re 16 g�n (aral�k: 1 g�n – 12,5 ay) olmu�tur. Medyan s�re 22 g�n (aral�k: 6 g�n ila 14,5+ ay; +, sans�rlenmi� bir de�eri g�sterir) olmu�tur.
Kortikosteroid kullan�m�n� gerektiren menenjit TECENTRIQ tedavisi g�ren hastalar�n %0,2'sinde (10/4.349) meydana gelmi�tir ve 8 (%0,2) hastada TECENTRIQ'in b�rak�lmas�na yol a�m��t�r.
�mm�nite ile ili�kili n�ropatiler:
Guillain-Barré sendromu ve demiyalizan polin�ropati, TECENTRIQ monoterapisi g�ren hastalar�n %0,1'inde (6/4.349) meydana gelmi�tir. Ba�lang�ca kadar ge�en medyan s�re 4,1 ay (aral�k: 17 g�n – 8,1 ay) olmu�tur. Medyan s�re 8 ay (aral�k: 19 g�n – 24,5 ay+, "+" sans�rlenmi� bir de�eri g�sterir). Bir (<%0,1) hasta, Guillain-Barré sendromu nedeniyle TECENTRIQ kullan�m�n� b�rakm��t�r. Kortikosteroid kullan�m� gerektiren Guillain-Barré sendromu TECENTRIQ tedavisi g�ren hastalar�n %0,1'inden az�nda (3/4.349) meydana gelmi�tir.
Miyastenik sendrom:
Miyestenia gravis, TECENTRIQ monoterapisi g�ren hastalar�n <%0,1'inde (1/4.349) meydana gelmi�tir. Ba�lang�ca kadar ge�en s�re 1,2 ayd�r.
�mm�nite ile ili�kili pankreatit:
Y�ksek amilaz ve y�ksek lipaz dahil olmak �zere pankreatit, TECENTRIQ monoterapisi g�ren hastalar�n %0,7'sinde (32/4.349) meydana gelmi�tir. Ba�lang�ca kadar ge�en medyan s�re 5,5 ay (aral�k: 1 g�n – 24,8 ay) olmu�tur. Medyan s�re 24 g�n (aral�k: 3+ g�n – 22,4+ ay; "+" sans�rlenmi� bir de�eri g�sterir) olmu�tur. Pankreatit, 3 hastada (<%0,1) TECENTRIQ'in b�rak�lmas�na yol a�m��t�r. Kortikosteroid kullan�m� gerektiren pankreatit olgular� TECENTRIQ monoterapisi g�ren hastalar�n %0,1'inde (5/4.349) meydana gelmi�tir.
�mm�nite ile ili�kili miyokardit:
Miyokardit, TECENTRIQ monoterapisi g�ren hastalar�n <%0,1'inde (3/4.349) meydana gelmi�tir. Ba�lang�ca kadar ge�en medyan s�re 2,1 ay (aral�k: 1,5 ay – 4,9 ay) olmu�tur. Medyan s�re 14 g�n (aral�k: 14 g�n – 2,8 ay) olmu�tur. �ki hastada (<%0,1) kortikosteroid kullan�m� gerekmi�tir ve 2 (<%0,1) hastada TECENTRIQ tedavisi durdurulmu�tur.
�mm�nite ile ili�kili nefrit:
Nefrit, TECENTRIQ tedavisi g�ren hastalar�n %0,2'sinde(10/4.349) meydana gelmi�tir. Ba�lang�ca kadar ge�en medyan s�re 5 ay (aral�k: 2 g�n – 17,5 ay) olmu�tur. Nefrit 5 hastada (%0,1) TECENTRIQ'in b�rak�lmas�na neden olmu�tur. D�rt hastada (<%0,1) kortikosteroid kullan�m�na gerek duyulmu�tur.
�mm�nite ile ili�kili miyozit:
Miyozit, TECENTRIQ monoterapisi g�ren hastalar�n %0,5'inde (20/4.349) meydana gelmi�tir. Ba�lang�ca kadar ge�en medyan s�re 3,3 ayd�r (aral�k: 12 g�n - 11 ay). Medyan s�re 5,7 ay (aral�k: 2 g�n - 36,9 +ay; + sans�rlenmi� bir de�eri g�sterir). Miyozit 2 hastada (<%0,1) TECENTRIQ'in b�rak�lmas�na neden olmu�tur. 7 hastada (%0,2) kortikosteroid kullan�m�na gerek duyulmu�tur.
�mm�nite ile ili�kili �iddetli kutan�z advers reaksiyonlar:
TECENTRIQ monoterapisi g�ren hastalar�n %0,6's�nda (28/4.349) �iddetli kutan�z advers reaksiyonlar meydana gelmi�tir. 28 hastadan birinde �l�mc�l olay meydana gelmi�tir. Ba�lang�ca kadar ge�en medyan s�re 5,2 ayd�r (aral�k: 4 g�n - 15,5 ay). Medyan s�re 2,4 ay (aral�k: 1 g�n - 37,5 +ay; + sans�rlenmi� bir de�eri g�sterir). �iddetli kutan�z advers reaksiyonlar 3 hastada (< %0,1) TECENTRIQ tedavisinin b�rak�lmas�na neden olmu�tur. %0,2 hastada (9/4.349) kortikosteroid kullan�m�na gerek duyulmu�tur.
�mm�nojenite:
�oklu faz II ve III �al��malar�nda, hastalar�n %13,1 ile %54,1'i tedaviyleortaya ��kananti-ila� antikorlar� (ADA) geli�tirmi�tir. Tedavi sonucu olu�mu� anti-ila� antikoru (ADA) geli�en hastalar�n ba�lang��ta genel olarak sa�l�k durumu ve hastal�k karakteristikleri a��s�ndan daha zay�f oldu�u g�r�lm��t�r. Ba�lang��taki bu sa�l�k ve hastal�k karakteristiklerindeki dengesizlikler, farmakokinetik, etkililik ve g�venlilik analizlerinin yorumlanmas�nda kar���kl�k yaratabilmektedir. Anti-ila� antikorlar�n�n (ADA) etkilili�e etkisini ara�t�rmak i�in ba�lang��taki sa�l�k ve hastal�k karakteristiklerindeki dengesizlikleri ayarlayan ke�if analizleri yap�lm��t�r. Bu analizlerde ADA geli�tiren hastalar�n, ADA geli�tirmeyen hastalara k�yasla etkililik faydas�nda azalma olas�l��� g�zard� edilmemi�tir. Anti-ila� antikorlar�n�n ba�lang�ca kadar ge�en medyan s�resi 3 ila 5 hafta olmu�tur.
TECENTRIQ monoterapisi (N=3.460) ve kombinasyon tedavisi (N=2.285) g�ren hastalardan elde edilen hasta havuzu verilerinden, ADA-pozitif pop�lasyonuna kar�� ADA-negatif pop�lasyonundan elde edilen advers olaylar�n s�kl��� s�ras�yla: Monoterapi i�in; 3. ve 4. derece yan etkiler %42,6'ya kar�� %39,4, ciddi yan etkiler %39,6'ya kar�� %33,3, tedavinin kesilmesine neden olan yan etkiler %8,5'e kar�� %7,8 iken; Kombinasyon tedavisi i�in 3. ve 4. derece yan etkiler, %63,9'a kar�� %60,9, ciddi yan etkiler %43,9'a kar�� 35,6, tedavinin kesilmesine neden olan yan etkiler % 22,8'e kar�� %18,4 olmu�tur. Ancak mevcut verilerden
yola ��karak olas� ila� advers reaksiyonlar�n�n yola�� hakk�nda kesin sonu�lara var�lamamaktad�r.
Pediyatrik pop�lasyon
TECENTRIQ'in �ocuklar ve ad�lesanlardaki g�venlili�i bilinmemektedir. 69 pediyatrik hastada (<18 ya�) yap�lan bir klinik �al��mada yeni bir g�venlilik sinyali olu�mam��t�r ve g�venlilik profili eri�kinlerinki ile kar��la�t�r�labilirdir.
Geriyatrik pop�lasyon
TECENTRIQ monoterapisi g�ren 65 ya� ve �zerindeki hastalar ile daha gen� hastalar aras�nda genel olarak bir g�venlilik farkl�l��� g�zlemlenmemi�tir.
��pheli advers reaksiyonlar�n raporlanmas�
Ruhsatland�rma sonras� ��pheli ila� advers reaksiyonlar�n�n raporlanmas� b�y�k �nem ta��maktad�r. Raporlama yap�lmas�, ilac�n yarar/risk dengesinin s�rekli olarak izlenmesine olanak sa�lar. Sa�l�k mesle�i mensuplar�n�n herhangi bir ��pheli advers reaksiyonu T�rkiye Farmakovijilans Merkezi (T�FAM)'ne bildirmeleri gerekmektedir (www.titck.gov.tr; e-posta: tufam@titck.gov.tr; tel: 0 800 314 00 08; faks:0 312 218 35 99).
4.9. Doz a��m� ve tedavisi
Atezolizumab doz a��m�na ili�kin bilgi mevcut de�ildir.
Doz a��m� durumunda, hastalar advers reaksiyon belirtileri veya semptomlar� bak�m�ndan yak�ndan izlenmeli ve uygun semptomatik tedavi ba�lat�lmal�d�r.
5. FARMAKOLOJ�K �ZELL�KLER
5.1. Farmakodinamik �zellikler
Farmakoterap�tik grup: Antineoplastik ve �mm�nomod�lat�r ajanlar, Monoklonal Antikorlar ve Antik�r �la� Konjugatlar�, PD-1/PDL-1 (Programlanm�� h�cre �l�m proteini 1 / �l�m ligand� 1) �nhibit�rleri
ATC kodu: L01FF05
Etki mekanizmas�:
H�manize IgG1 anti-programl� �l�m-ligand� 1(PD-L1), t�m�r h�creleri ve/veya t�m�r infiltre eden imm�n h�crelerinde eksprese olabilir ve t�m�r mikroortam�nda anti-t�m�r imm�n yan�t�n�n inhibisyonuna katk�da bulunabilir. PD-L1'in T-h�crelerinde ve antijen sunan h�crelerde bulunan PD-1 ve B7.1 resept�rlerine ba�lanmas�, sitotoksik T-h�cre aktivitesini, T-h�cre �o�almas�n� ve sitokin �retimini bask�lar.
Atezolizumab Fc b�lgesi de�i�tirilmi� bir h�manize imm�noglob�lin G1 (IgG1) monoklonal antikorudur; do�rudan PD-L1'e ba�lan�r ve PD-1 ve B7.1 resept�rlerinin ikili blokaj�n� sa�layarak, antikor ba��ml� h�cresel sitotoksisiteyi ind�klemeden antit�m�r imm�n yan�t�n yeniden aktive edilmesi de dahil, imm�n yan�t�n PD-L1/PD-1 arac�l� inhibisyonunu serbest b�rak�r. Atezolizumab, PD-L2/PD-1 etkile�imini koruyarak PD-L2/PD-1 arac�l� inhibit�r
sinyallerin devam etmesine izin verir.
Klinik etkililik ve g�venlilik:
1200 mg dozda �� haftada bir uygulanan TECENTRIQ ile yap�lan �al��malar�n tan�m� i�in, TECENTRIQ 1200 mg/20 mL inf�zyonluk ��zelti haz�rlamak i�in konsantre'nin K�sa �r�n Bilgisi'ne bak�n�z.
��l� negatif meme kanseri
IMpassion130 (WO29522): Daha �nce metastatik hastal�k i�in tedavi edilmemi�, lokal ileri veya metastatik �NMK hastalar�nda randomize faz III �al��ma
Metastatik hastal�k i�in daha �nce kemoterapi almam��, rezeke edilemeyen, lokal ileri veya metastatik �NMK olan hastalarda, atezolizumab ile nab-paklitakselin etkililik ve g�venlili�ini de�erlendirmek i�in Faz III, �ift k�r, iki kollu, �ok merkezli, uluslararas�, randomize, plasebo kontroll� bir �al��ma olan IMpassion130 y�r�t�lm��t�r. Taksan monoterapisi i�in uygun olan hastalar se�ilmi�tir (yani h�zl� klinik ilerlemenin olmamas�, ya�am� tehdit eden viseral metastazlar�n veya h�zl� semptom ve/veya hastal�k kontrol� ihtiyac�n�n olmamas�). Son 12 ay i�erisinde neoadjuvan veya adjuvan ortam�nda kemoterapi alan hastalar, otoimm�n hastal�k �yk�s� olan, randomizasyondan �nceki 4 hafta i�inde bir canl� aten�e a��, randomizasyondan �nceki 4 hafta i�inde sistemik imm�nostim�lat�r ajan uygulanm�� veya randomizasyondan �nceki 2 hafta i�inde sistemik immunosupresif t�bbi �r�n kullanm�� hastalar, tedavi edilmemi� semptomatik veya kortikosteroide ba�l� beyin metastaz� �yk�s� olan hastalar dahil edilmemi�tir. T�m�r de�erlendirmeleri, 1. d�ng�n�n (1. g�n) ard�ndan ilk 12 ay s�reyle 8 haftada (± 1 hafta) bir ve sonras�nda 12 haftada (± 1 hafta) bir olmak �zere ger�ekle�tirilmi�tir.
Toplamda 902 hasta �al��maya dahil edilmi� ve karaci�er metastaz� varl���, taksan tedavisi �yk�s� ve t�m�r infiltre edici imm�n h�crelerde (IC) PD-L1 ekspresyonu durumuna g�re tabakaland�r�lm��t�r (PD-L1 ile boyanm�� t�m�r infiltre edici imm�n h�creler [IC] < %1 VENTANA PD-L1 (SP142) testi ile de�erlendirilen t�m�r alan�na kar�� t�m�r alan�n�n ≥ %1'i).
Hastalar, her 28 g�nl�k siklusta 1. ve 15. g�nlerde intraven�z inf�zyon yoluyla atezolizumab 840 mg veya plasebo ve her 28 g�nl�k siklusta 1., 8. ve 15. g�nlerde intraven�z inf�zyon yoluyla nab-paklitaksel (100 mg/m) almak �zere randomize edilmi�tir. Hastalar, RECIST v1.1 uyar�nca radyografik hastal�kta ilerleme veya kabul edilemez bir toksisite meydana gelene kadar tedavi g�rm��t�r. Kabul edilemez toksisite nedeniyle nab-paklitaksel durduruldu�unda atezolizumab ile tedaviye devam edilebilir. Her tedavi kolunda medyan tedavi d�ng�s� say�s� atezolizumab i�in 7 ve nab-paklitaksel i�in 6'd�r.
�al��ma pop�lasyonunun demografik ve ba�lang�� �zellikleri, tedavi kollar� aras�nda iyi dengelenmi�tir. Hastalar�n �o�u kad�nd�r (%99,6), %67,5'i beyaz, %17,8'i Asyal�d�r. Medyan ya� 55' dir (aral�k: 20-86). Ba�lang��ta ECOG performans skoru 0 (%58,4) veya 1'dir (%41,3). Genel olarak, dahil edilen hastalar�n %41'inde ba�lang��ta PD-L1 ekspresyonu ≥ %1 iken, %27'sinde karaci�er metastazlar� ve %7'sinde beyin metastazlar� mevcuttur. Yakla��k olarak hastalar�n yar�s�, (neo)adjuvan ko�ullarda bir taksan (%51) veya antrasiklin (%54) alm��t�r. PD-L1 ekspresyonu ≥ %1 olan hastalarda demografik bilgiler ve ba�lang�� t�m�r hastal���, genel itibariyle daha geni� �al��ma pop�lasyonunu temsil ediyordu.
E� birincil etkililik sonlan�m noktalar�, ITT pop�lasyonunda ve RECIST v1.1 uyar�nca PD-L1 ekspresyonu ≥ %1 olan hastalarda ara�t�rmac� taraf�ndan de�erlendirilen, progresyonsuz sa�kal�m�n (PS) yan� s�ra ITT pop�lasyonunda ve PD-L1 ekspresyonu ≥ %1 olan hastalarda
genel sa�kal�m� (GS) i�ermi�tir. �kincil etkililik sonlan�m noktalar�, RECIST v1.1 uyar�nca objektif yan�t oran� (OYO) ve yan�t s�resini (YS) i�ermi�tir.
Medyan sa�kal�m takibi 13 ayl�k olan progresyonsuz sa�kal�m (PS) i�in son analiz s�ras�nda PD-L1 ekspresyonu ≥%1 olan hastalar i�in IMpassion130'un PS, OYO ve YS sonu�lar� Tablo 3'de �zetlenmi�tir ve PS i�in Kaplan-Meier e�rileri PD-L1 ekspresyonu <%1 olan hastalar �ekil 1'de g�sterilmektedir. Nab-paklitaksele atezolizumab eklendi�inde PS'de iyile�me g�zlenmemi�tir (TO 0,94, %95 GA 0,78, 1,13).
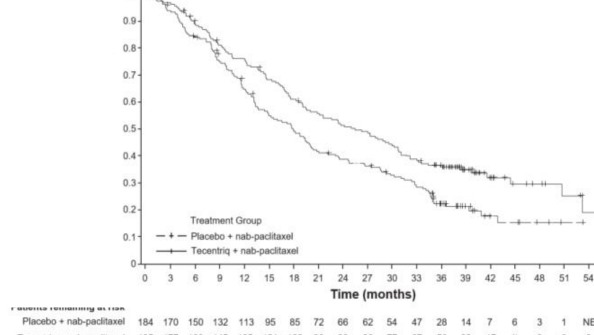
Nihai GS analizi, PD-L1 ekspresyonu ≥%1 olan ve medyan takip s�resi 19,12 olan hastalarda yap�lm��t�r. GS sonu�lar� Tablo 3'de ve Kaplan-Meier e�rileri �ekil 1'de sunulmaktad�r. PD-L1 ekspresyonu <%1 olan hastalar, nab-paklitaksele atezolizumab eklendi�inde geli�mi� GS g�zlenmemi�tir (TO 1,02, %95 GA 0,84, 1,24).
PD-L1 ekspresyonu ≥%1 olan hastalarda, �nceki (neo)adjuvan tedavi, BRCA1/2 mutasyonu ve ba�lang��ta asemptomatik beyin metastazlar� ara�t�r�larak ke�if ama�l� alt grup analizleri yap�lm��t�r. Daha �nce (neo) adjuvan tedavi alm�� hastalarda (n=242) birincil (nihai) PS i�in tehlike oran� 0,79 ve final GS i�in 0,77 iken, daha �nce (neo)adjuvan tedavi almam�� hastalarda (n=127), birincil (nihai) PS i�in tehlike oran�, final GS i�in 0,44 ve 0,54 olarak saptanm��t�r.
IMpassion130 �al��mas�nda, test edilen 614 hastan�n 89'u (%15) patojenik BRCA1/2 mutasyonlar� ta��maktayd�. PD-L1+/BRCA1/2 mutant alt grubundan 19 hasta atezolizumab art� nab-paklitaksel ve 26 plasebo art� nab-paklitaksel alm��t�r. Ara�t�rmac� analize dayanarak ve k���k �rnek boyutunun kabul edilmesiyle, BRCA1/2 mutasyonunun varl���, atezolizumab ve nab-paklitakselin PS klinik yarar�n� etkilemiyor gibi g�r�nmektedir.
Tedavi edilen hasta say�s� az olmas�na ra�men, ba�lang��ta asemptomatik beyin metastaz� olan hastalarda etkililik kan�t� yoktu; medyan PS, atezolizumab art� nab-paklitaksel kolunda (n=15) 2,2 ay iken, plasebo art� nab-paklitaksel kolunda (n=11) 5,6 ayd�r (TO 1,40; %95 GA 0,57,
3,44).
Tablo 3: PD-L1 ekspresyonu ≥ %1 olan hastalarda etkililik �zeti (IMpassion130)
Anahtar etkililik sonlan�m noktalar� | Atezolizumab+ nab-paklitaksel | Plasebo+ nab-paklitaksel |
Birincil etkililik sonlan�m noktalar� | n=185 | n=184 |
Ara�t�rmac� taraf�ndan de�erlendirilen PS (RECIST v1.1)-Primer analiz | ||
Olay say�s� (%) | 138 (%74,6) | 157 (%85,3) |
Medyan PS s�resi (ay) | 7,5 | 5 |
%95 GA | (6,7; 9,2) | (3,8; 5,6) |
Tabakaland�r�lm�� tehlike oran�‡ (%95 GA) | 0,62 (0,49; 0,78) | |
p de�eri | <0,0001 | |
12-ayl�k PS (%) | 29,1 | 16,4 |
Ara�t�rmac� taraf�ndan de�erlendirilen PS (RECIST v1.1)- G�ncellenmi� ke�if analizi | ||
Olay say�s� (%) | 149 (%80,5) | 163 (%88,6) |
Medyan PS s�resi (ay) | 7,5 | 5,3 |
%95 GA | (6,7; 9,2) | (3,8; 5,6) |
Tabakaland�r�lm�� tehlike oran�‡ (%95 GA) | 0,63 (0,5; 0,8) | |
p de�eri | <0,0001 | |
12-ayl�k PS (%) | 30,3 17,3 | |
GS | ||
�l�m say�s�(%) | 120 (%64,9) | 139 (%75,5) |
Medyan olay s�resi (ay) | 25,4 | 17,9 |
%95 GA | (19,6, 30,7) | (13,6, 20,3) |
Tabakaland�r�lm�� tehlike oran�‡ (%95 GA) |
|
0,67 (0,53, 0,86) |
�kincil ve ara�t�rmaya y�nelik sonlan�m noktalar� | ||
Ara�t�rmac� taraf�ndan de�erlendirilen OYO (RECIST 1.1) | n=185 | n=183 |
Yan�t veren hasta say�s� (%) | 109 (%58,9) | 78 (%42,6) |
%95 GA | (51,5; 66,1) | (35,4; 50,1) |
Tam yan�t say�s� (%) | 19 (%10,3) | 2 (%1,1) |
K�smi yan�t say�s� (%) | 90 (%48,6) | 76 (%41,5) |
Stabil hastal�k say�s� | 38 (%20,5) | 49 (%26,8) |
Ara�t�rmac� taraf�ndan de�erlendirilen YS | n=109 | n=78 |
Ay olarak medyan | 8,5 | 5,5 |
%95 GA | (7,3; 9,7) | (3,7; 7,1) |
‡ Karaci�er metastaz� varl��� ve taksan tedavisi �yk�s�ne g�re tabakaland�r�lm��t�r.
PS= Progresyonsuz sa�kal�m; RECIST= Solid T�m�rlerde Yan�t De�erlendirme Kriterleri v1.1.; GA= G�ven aral���; OYO= Objektif yan�t oran�; YS= Yan�t s�resi; GS= Genel sa�kal�m
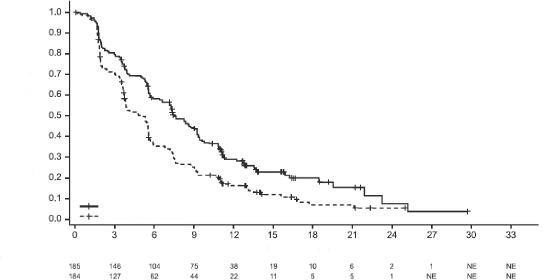
�ekil 1: PD-L1 ekspresyonu ≥ %1 olan hastalarda Progresyonsuz Sa�kal�m i�in Kaplan-Meier E�risi (IMpassion130)
�ekil 2: PD-L1 ekspresyonu ≥ %1 olan hastalarda Genel Sa�kal�m i�in Kaplan-Meier E�risi (IMpassion130)
EORTC QLQ-C30 ile �l��len hasta taraf�ndan bildirilen genel sa�l�k durumu/sa�l�kla ili�kili ya�am kalitesinin (HRQoL) bozulmas�na kadar ge�en s�re (ba�lang�� puan�ndan s�rekli ≥10 puanl�k bir d����) her tedavi grubunda benzerdir ve bu t�m hastalar�n kar��la�t�r�labilir bir s�re boyunca temel HRQoL'lerini korudu�unu g�stermektedir.
5.2. Farmakokinetik �zellikler
Genel �zelliklerAtezolizumaba maruziyet 1 mg/kg - 20 mg/kg doz aral���nda 3 haftada bir uygulanan sabit doz
1200 mg doz ile orant�l� olarak artm��t�r. 472 hastay� i�eren bir pop�lasyon analizi, a�a��daki doz aral��� i�in atezolizumab farmakokineti�ini birinci derece eliminasyonla bir do�rusal iki b�lmeli d�zenleme modeli ile 1 - 20 mg/kg olarak a��klam��t�r. �� haftada bir uygulanan 1200 mg atezolizumab dozu ve 2 haftada bir uygulanan 840 mg atezolizumab dozu ve 4 haftada bir uygulanan 1680 mg atezolizumab dozunun farmakokinetik �zellikleri ayn�d�r. Bu �� doz rejimiyle kar��la�t�r�labilir toplam maruziyetlere ula��lmas� beklenmektedir. Bir pop�lasyon farmakokinetik analizi, 6-9 hafta tekrarl� dozlamadan sonra kararl� durumun elde edildi�ini �ne s�rmektedir. E�ri alt�ndaki alanda, maksimum konsantrasyon ve en d���k konsantrasyonda sistemik birikim s�ras�yla 1,91; 1,46 ve 2,75 kat olmu�tur.
Emilim:
Atezolizumab intraven�z inf�zyon �eklinde uygulan�r. Di�er uygulama yollar�yla yap�lan �al��malar olmam��t�r.
Da��l�m:
Pop�lasyon farmakokinetik analizi, bir hastada merkezi kompartman da��l�m hacminin 3,28 L ve kararl� durumunda hacmin 6,91 L oldu�unu g�stermektedir.
Biyotransformasyon:
Atezolizumab�n metabolizmas� do�rudan ara�t�r�lmam��t�r. Antikor klerensi esas olarak katabolizmayla ger�ekle�ir.
Eliminasyon:
Pop�lasyon farmakokinetik analizi, atezolizumab�n klerensinin 0,2 L/g�n ve tipik terminal eliminasyon yar� �mr�n�n 27 g�n oldu�unu g�stermektedir.
�zel pop�lasyonlara ili�kin ek bilgiler:
Pop�lasyon farmakokineti�i ve maruziyet-yan�t analizlerine g�re a�a��daki fakt�rlerin atezolizumab�n farmakokineti�i �zerinde bir etkisi yoktur: Ya�, (21-89 ya�), b�lge, etnik k�ken, b�brek bozuklu�u, hafif karaci�er bozuklu�u, PD-L1 ekspresyonu d�zeyi veya ECOG performans durumu. V�cut a��rl���, cinsiyet, pozitif ADA durumu, alb�min seviyeleri ve t�m�r y�k�n�n atezolizumab farmakokineti�i �zerindeki etkisi istatistiki olarak anlaml� ancak klinik olarak anlaml� de�ildir. Doz ayarlamas� �nerilmemektedir.
Pediyatrik pop�lasyon:
Pediyatrik (<18 ya�, n=69) ve gen� eri�kin (18-30 ya�, n=18) hastalarda y�r�t�len erken faz, �ok merkezli, a��k etiketli bir �al��madan elde edilen farmakokinetik sonu�lar, atezolizumab klerensi ve da��l�m hacminin, normal v�cut a��rl���na g�re normalize edildi�inde 15 mg/kg alan pediyatrik hastalar ve her 3 haftada bir 1200 mg atezolizumab alan gen� eri�kin hastalar aras�nda kar��la�t�r�labilir oldu�unu g�stermi�tir. Maruziyetin ise, v�cut a��rl��� d��t�k�e pediyatrik hastalarda artt��� g�zlenmi�tir. Bu de�i�iklikler, atezolizumab konsantrasyonunun terap�tik hedef maruziyetinin alt�na d��mesi ile ba�lant�l� de�ildir. 2 ya� alt�ndaki �ocuklar i�in veriler s�n�rl�d�r, bu nedenle kesin sonu�lara var�lamamaktad�r.
Geriyatrik pop�lasyon:
Ya�l� hastalarda TECENTRIQ i�in �zel bir �al��ma yap�lmam��t�r. Ya��n atezolizumab�n farmakokineti�i �zerindeki etkisi bir pop�lasyon farmakokinetik analizinde de�erlendirilmi�tir. Ya�, 21-89 ya� aral���ndaki (n=472) ve medyan 62 ya��ndaki hastalar temel al�nd���nda, atezolizumab�n farmakokineti�ini etkileyen �nemli bir kovaryant olarak tan�mlanmam��t�r.
<65 ya��ndaki (n=274), 65-75 ya��ndaki (n=152) ve >75 ya��ndaki (n=46) hastalarda atezolizumab�n farmakokineti�inde klinik a��dan anlaml� bir fark g�zlenmemi�tir (bkz. B�l�m 4.2).
B�brek yetmezli�i:
B�brek yetmezli�i olan hastalarda TECENTRIQ i�in �zel bir �al��ma yap�lmam��t�r. Pop�lasyon farmakokinetik analizinde, b�brek fonksiyonu normal (90 mL/dk/1,73 m veya �zeri tahmini glomer�ler filtrasyon h�z� [eGFR]; n=140) olan hastalarla kar��la�t�r�ld���nda, hafif (60 - 89 mL/dk/1,73 m eGFR; n=208) veya orta �iddetli (30 - 59 mL/dk/1,73 m eGFR; n=116) b�brek yetmezli�i olan hastalarda atezolizumab�n klerensinde klinik a��dan �nemli farklar bulunmam��t�r. Yaln�zca birka� hastada �iddetli b�brek yetmezli�i vard�r (eGFR 15 - 29 mL/dk/1,73 m; n=8) (bkz. B�l�m 4.2). �iddetli b�brek yetmezli�inin atezolizumab�n farmakokineti�i �zerindeki etkisi bilinmemektedir.
Karaci�er yetmezli�i:
Karaci�er bozuklu�u olan hastalarda TECENTRIQ i�in �zel bir �al��ma yap�lmam��t�r. Pop�lasyon farmakokinetik analizinde, hafif (bilirubin ≤N�S ve AST>N�S veya bilirubin >1 – 1,5 x N�S ve herhangi bir AST) veya orta (bilirubin > 1,5 – 3 x N�S ve herhangi bir AST) karaci�er yetmezli�i olan ve normal karaci�er fonksiyonu (bilirubin ≤ N�S ve AST ≤N�S) olan hastalar aras�nda atezolizumab�n klerensi bak�m�ndan klinik olarak �nemli farklar bulunmam��t�r. �iddetli (bilirubin > 3 x N�S ve herhangi bir AST) karaci�er yetmezli�i olan hastalara ili�kin veri mevcut de�ildir. Karaci�er yetmezli�i, Ulusal Kanser Enstit�s� (NCI) karaci�er fonksiyon bozuklu�u kriterlerine g�re tan�mlanm��t�r (bkz. B�l�m 4.2). �iddetli karaci�er yetmezli�inin (bilirubin > 3 x N�S ve herhangi bir AST) atezolizumab�n farmakokineti�i �zerindeki etkisi bilinmemektedir.
5.3. Klinik �ncesi g�venlilik verileri
Karsinojenite:
TECENTRIQ'in karsinojenik potansiyelini belirlemek i�in karsinojenite �al��mas� yap�lmam��t�r.
Mutajenite:
TECENTRIQ'in mutajenik potansiyelini belirlemek i�in mutajenite �al��mas� yap�lmam��t�r. Bununla birlikte, monoklonal antikorlar�n DNA veya kromozomlar� de�i�tirmesi beklenmemektedir.
Fertilite:
TECENTRIQ ile herhangi bir do�urganl�k �al��mas� yap�lmam��t�r. Bununla birlikte, kronik toksisite �al��mas�na sinomolgus maymunlar�nda erkek ve di�i �reme organlar�n�n de�erlendirilmesi dahil edilmi�tir. Atezolizumab�n di�i maymunlara tahmini EAA'da uygulanmas� (�nerilen dozu alan hastalardaki EAA'n�n yakla��k 6 kat�), geri d�n���ml� olarak d�zensiz adet d�ng�s�ne ve yumurtal�klarda yeni olu�turulmu� korpus lutea eksikli�ine neden olmu�tur. Erkek �reme organlar� �zerinde herhangi bir etkisi olmam��t�r.
Teratojenite:
TECENTRIQ ile hayvanlarda �reme ve teratojenite �al��malar� yap�lmam��t�r. Hayvan �al��malar�, PD-L1/PD-1 yola��n�n inhibisyonunun, geli�en fet�s�n ba����kl�kla ili�kili reddine yol a�arak fetal �l�mle sonu�lanabilece�ini g�stermi�tir. TECENTRIQ uygulamas�, embriyo-fetal �l�m dahil olmak �zere fetal zarara neden olabilir.
6. FARMAS�T�K �ZELL�KLER
6.1. Yard�mc� maddelerin listesi
L-histidin
Glasiyal asetik asit S�kroz
Polisorbat 20 Enjeksiyonluk su
6.2. Ge�imsizlikler
Bu t�bbi �r�n, B�l�m 6.6'da belirtilenlerin d���nda di�er t�bbi �r�nlerle kar��t�r�lmamal�d�r.
6.3. Raf �mr�
A��lmam�� flakon: 36 ay
Seyreltilmi� ��zelti: Haz�rlama zaman�ndan sonra 2-8°C'de 24 saat ve ortam s�cakl���nda (≤30°C) 24 saat i�inde kullan�mdaki kimyasal ve fiziksel stabilite g�sterilmi�tir.
Mikrobiyolojik a��dan, haz�rlanan inf�zyonluk ��zelti hemen kullan�lmal�d�r. Hemen kullan�lmazsa, kullan�m s�ras�ndaki saklama s�releri ve kullan�m �ncesi ko�ullar kullan�c�n�n sorumlulu�undad�r ve normalde 2-8°C'de 24 saatten fazla veya ortam s�cakl���nda (≤25°C) 8 saatten fazla olmamal�d�r.
6.4. Saklamaya y�nelik �zel tedbirler
Buzdolab�nda saklay�n�z (2°C-8°C).
I��ktan korumak i�in flakonu karton kutusunda saklay�n�z. Dondurmay�n�z. �alkalamay�n�z.
T�bbi �r�n�n seyreltme sonras�nda saklama ko�ullar� i�in bkz. B�l�m 6.3.
6.5. Ambalaj�n niteli�i ve i�eri�i
14 mL ��zelti i�eren tapal� (butil kau�uk) flakon (Tip I cam). Bir flakonluk paket.
6.6. Be�eri t�bbi �r�nden arta kalan maddelerin imhas� ve di�er �zel �nlemler
TECENTRIQ herhangi bir antimikrobiyal koruyucu i�ermez ve bir sa�l�k meslek mensubu taraf�ndan aseptik teknik kullan�larak haz�rlanmal�d�r.
Seyreltme talimatlar�:
14 mL TECENTRIQ konsantresi flakondan �ekilmeli ve i�inde sodyum klor�r 9 mg/mL (% 0,9) enjeksiyonluk ��zelti bulunan 250 mL'lik bir PVC, polietilen (PE) veya poliolefin inf�zyon torbas� i�ine seyreltilmelidir. Seyreltmeden sonra ��zeltinin bir mL'si yakla��k 3,2 mg TECENTRIQ (840 mg/264 mL) i�ermelidir. Torba, k�p�k olu�umuna izin vermeden ��zeltiyi kar��t�rmak i�in yava��a alt �st edilmelidir. �nf�zyon haz�rland���nda hemen uygulanmal�d�r (bkz. B�l�m 6.3).
Parenteral t�bbi �r�nler uygulanmadan �nce partik�ller ve renk de�i�imi a��s�ndan ��plak g�zle incelenmelidir. Partik�ller veya renk de�i�imi g�zlenirse ��zelti kullan�lmamal�d�r.
TECENTRIQ ile �r�ne temas eden polivinil klor�r (PVC), polietilen (PE) veya poliolefin (PO) y�zeyleri olan intraven�z torbalar aras�nda ge�imsizlik g�zlenmemi�tir. �lave olarak,
polieters�lfon veya polis�lfon i�eren d�z eksenli filtre membranlar� ve inf�zyon setleri ile PVC, PE, polibutadien veya polieter�retan i�eren di�er inf�zyon yard�mc�lar� ile de ge�imsizlik g�zlenmemi�tir. D�z eksenli filtre membranlar�n�n kullan�lmas� se�ime ba�l�d�r.
Kullan�lmam��/son kullanma tarihi ge�mi� ila�lar�n imhas�:
T�bbi �r�nlerinin �evreye sal�nmas� en aza indirilmelidir. �la�lar, at�k suyla birlikte at�lmamal�d�r ve evsel at�klarla imhas�ndan ka��n�lmal�d�r.
Kullan�lmam�� olan �r�nler ya da at�k materyaller, “T�bbi At�klar�n Kontrol� Y�netmeli�i'' ve “Ambalaj At�klar�n�n Kontrol� Y�netmeli�i”ne uygun olarak imha edilmelidir.
 HIV ve Aids
HIV, Human Immunodeficiency Virus’d�r (�nsanlarda Ba����kl�k Sistemini Bozan
Vir�sd�r). Bu vir�s AIDS hastal���na sebep olur.
HIV ve Aids
HIV, Human Immunodeficiency Virus’d�r (�nsanlarda Ba����kl�k Sistemini Bozan
Vir�sd�r). Bu vir�s AIDS hastal���na sebep olur. |
 Mesane Kanseri
Mesane kanseri her zaman mukozada ba�lar. Erken safhalarda bu tabakada s�n�rl� kal�r ve
h�cre i�indeki karsinom olarak nitelendirilir.
Mesane Kanseri
Mesane kanseri her zaman mukozada ba�lar. Erken safhalarda bu tabakada s�n�rl� kal�r ve
h�cre i�indeki karsinom olarak nitelendirilir. |
 |
Ast�m Ast�ml� ki�ilerin akci�erlerindeki hava borular� (bron�lar) hassast�r. Bu ki�iler belirli tetikleyici fakt�rlere maruz kald�klar�nda, hava borular� nefes almalar�n� g��le�tirecek �ekilde daral�r. |
 |
Do�um Sonras� Depresyonu Do�um sonras� depresyonu, do�umdan sonra her on kad�ndan biri taraf�ndan tecr�be edilen stresli bir durumdur. |
 |
L�semi Kan Kanseri L�semi, kan kanseridir ve v�cudunun kan olu�turan dokular�n�n hastalanmas� anlam�na gelir. Bir�ok l�semi t�r� vard�r; baz� l�semi t�rleri �ocuklarda baz�lar� da yeti�kinlerde s�k g�r�l�r. |
�LA� GENEL B�LG�LER�
Roche M�stahzarlar� Sanayi A.�.
| Sat�� Fiyat� | 79822.29 TL [ 3 Aug 2026 ] |
| �nceki Sat�� Fiyat� | 79822.29 TL [ 27 Jul 2026 ] |
| Original / Jenerik | Original �la� |
| Re�ete Durumu | K�rm�z� Re�eteli bir ila�d�r. |
| Barkodu | 8699505763323 |
| Etkin Madde | Atezolizumab |
| ATC Kodu | L01XC32 |
| Birim Miktar | 840/14 |
| Birim Cinsi | MG/ML |
| Ambalaj Miktar� | 1 |
| Antineoplastik ve �mm�nomod�lat�r Ajanlar > Di�er Kanser �la�lar� |
| �thal ( ref. �lke : Litvanya ) ve Be�eri bir ila�d�r. |