TECENTRIQ 1200 mg/20 ml inf�zyonluk ��zelti haz�rlamak i�in konsantre K�sa �r�n Bilgisi
{ Atezolizumab }
1. BE�ER� TIBB� �R�N�N ADI
TECENTRIQ 1200 mg/20 ml inf�zyonluk ��zelti haz�rlamak i�in konsantre Steril
2. KAL�TAT�F VE KANT�TAT�F B�LE��M
Etkin madde
20 mL konsantre bir flakon i�inde 1200 mg atezolizumab i�erir.
Dil�syondan sonra 1 mL sol�syon yakla��k olarak 4,4 mg atezolizumab i�erir (bkz. B�l�m 6.6).
Atezolizumab, rekombinant DNA teknolojisiyle �in hamsteri yumurtal�k h�crelerinde �retilen, bir Fc b�lgesi de�i�tirilmi�, h�manize IgG1 anti-programl� �l�m-ligand� 1 (PD-L1) monoklonal antikorudur.
Yard�mc� maddeler
Yard�mc� maddeler i�in B�l�m 6.1'e bak�n�z.
3. FARMAS�T�K FORMU
�nf�zyonluk ��zelti haz�rlamak i�in steril konsantre Berrak, renksiz ila hafif sar�ms� s�v�
4. KL�N�K �ZELL�KLER
4.1. Terap�tik endikasyonlar
Erken Evre K���k H�creli D��� Akci�er Kanseri (EE-KHDAK)
TECENTRIQ, PD-L1 ekspresyonu t�m�r h�crelerinde (TC) ≥ %50 olan, yeti�kin Evre II-IIIA k���k h�creli d��� akci�er kanseri hastalar�nda, rezeksiyonu ve platin bazl� kemoterapiyi takiben, adjuvan tedavide monoterapi olarak endikedir. Non-skuam�z k���k h�creli d��� akci�er kanserlerinde EGFR ve ALK mutasyonlar�n�n negatif olmas� gereklidir. Hastal�k n�ks� veya kabul edilemez toksisite olmad�k�a maksimum 1 y�l s�reyle kullan�m� uygundur.
Metastatik K���k H�creli D��� Akci�er Kanseri (KHDAK)
TECENTRIQ'in, performans durumu ECOG 0-1 olan, EGFR, ALK, ROS negatif, semptomatik beyin metastaz� olmayan, lokal ileri ve/veya metastatik k���k h�creli d��� akci�er kanseri (KHDAK) nedeniyle daha �nce 1-2 basamak kemoterapi alm�� ve progresyon geli�mi�
hastalar�n tedavisinde tekrar progresyona kadar kullan�m� endikedir.
TECENTRIQ, EGFR mutant ya da ALK pozitif olmayan ve PD-L1 ekspresyonu t�m�r h�crelerinde (TC) ≥ %50 ya da t�m�r infiltre edici imm�n h�crelerde ≥ %10 olan, aktif beyin metastaz� olmayan, ECOG 0 veya 1 metastatik k���k h�creli d��� akci�er kanseri yeti�kin hastalar�n birinci basamak tedavisinde monoterapi olarak endikedir. Yass� h�creli KHDAK hastalar�nda EGFR ve/veya ALK durumunun belirlenmi� olmas� aranmaz.
TECENTRIQ, ECOG performans skoru 0-1 olan, yeterli kardiyak, renal ve hepatik fonksiyonlar� bulunan, aktif beyin metastaz� olmayan, EGFR ya da ALK mutasyonlar� bulunmayan ve e� zamanl� imm�ns�presif veya kortikosteroid tedavisi almayan metastatik yass� h�creli olmayan KHDAK hastalar�n�n birinci basamak tedavisinde, nab-paklitaksel ve karboplatinli kemoterapi rejimi ile kombine olarak progresyona kadar kullan�m� endikedir. Tedavi sonu progresyonda di�er PD-1 ve PD-L1 inhibit�rleri kullan�lamaz.
K���k H�creli Akci�er Kanseri (KHAK)
TECENTRIQ'in, karboplatin ve etoposidle kombine olarak yayg�n evre k���k h�creli akci�er kanseri olan yeti�kin hastalar�n birinci basamak tedavisinde kullan�m� endikedir.
Hepatosel�ler Kanser
TECENTRIQ'in bevacizumab ile kombine olarak, daha �nce sistemik tedavi g�rmemi�, ECOG performans durumu 0 ve 1 olan, Child-Pugh skoru A olan, metastatik veya lokorejyonel tedaviye uygun olmayan rezeke edilemeyen hepatosel�ler karsinomlu yeti�kin hastalar�n tedavisinde kullan�m� endikedir.
4.2. Pozoloji ve uygulama �ekli
TECENTRIQ, kanser tedavisinde deneyimli bir hekimin g�zetimi alt�nda uygulanmal�d�r. KHDAK hastalar�nda PD-L1 testi:
TECENTRIQ monoterapisi
Erken evre KHDAK ve birinci basamak metastatik KHDAK hastalar�n�n tedaviye uygunlu�u, valide edilmi� bir test ile teyid edilmi� PD-L1 t�m�r ekspresyonuna g�re yap�lmal�d�r (bkz. B�l�m 5.1).
Pozoloji/Uygulama s�kl��� ve s�resi:
TECENTRIQ monoterapisi
�nerilen TECENTRIQ dozu, �� haftada bir intraven�z yoldan uygulanan 1200 mg'd�r.
TECENTRIQ kombinasyon tedavisi
Non-skuamoz KHDAK – TECENTRIQ ile nab-paklitaksel ve karboplatin kombinasyonu
�nd�ksiyon faz�nda, 1. g�nde �nerilen TECENTRIQ dozu intraven�z yoldan uygulanan 1200
mg ve takiben s�ras�yla nab-paklitaksel ve karboplatindir. Nab-paklitaksel 8. ve 15. g�nlerde intraven�z yoldan uygulan�r. Bu rejim 4 veya 6 siklus boyunca her 3 haftada bir uygulan�r.
�nd�ksiyon faz�n�, kemoterapi olmadan, yaln�zca 3 haftada bir 1200 mg intraven�z yoldan TECENTRIQ uygulanan idame faz� takip eder.
KHAK – TECENTRIQ ile karboplatin ve etoposid kombinasyonu
�nd�ksiyon faz�nda, 1. g�nde �nerilen TECENTRIQ dozu intraven�z yoldan uygulanan 1200 mg ve takiben s�ras�yla karboplatin ve etoposiddir. Etoposid 2. ve 3. g�nlerde intraven�z yoldan uygulan�r. Bu rejim 4 siklus boyunca her 3 haftada bir uygulan�r.
�nd�ksiyon faz�n�, kemoterapi olmadan, yaln�zca 3 haftada bir 1200 mg intraven�z yoldan TECENTRIQ uygulanan idame faz� takip eder.
Hepatosel�ler Kanser (HSK)
TECENTRIQ ile bevacizumab kombinasyonu
�nerilen TECENTRIQ dozu 1200 mg'd�r ve bunu takiben 15 mg/kg bevacizumab ile �� haftada bir intraven�z yoldan uygulan�r.
Tedavi s�resi:
2. Basamak K���k H�creli D��� Akci�er Kanseri (KHDAK) ve Hepatosel�ler Kanser (HSK)
Klinik faydan�n kaybedilmesine (bkz. B�l�m 5.1) veya y�netilemeyen toksisiteye kadar hastalar�n TECENTRIQ ile tedavi edilmeleri �nerilmektedir.
Erken Evre K���k H�creli D��� Akci�er Kanseri
Hastal�k n�ks� veya kabul edilemez toksisite olmad�k�a hastalar�n 1 y�l boyunca TECENTRIQ ile tedavi edilmeleri �nerilmektedir. 1 y�ldan uzun s�ren tedavi s�resi �al���lmam��t�r.
1. Basamak K���k H�creli D��� Akci�er Kanseri (KHDAK) (TECENTRIQ monoterapi
Hastal�k progresyonuna veya y�netilemeyen toksisiteye kadar hastalar�n TECENTRIQ ile tedavi edilmeleri �nerilmektedir.
1. Basamak non-skuamoz K���k H�creli D��� Akci�er Kanseri (KHDAK) (TECENTRIQ ile nab-paklitaksel ve karboplatin kombinasyonu)
Hastal�k progresyonuna veya y�netilemeyen toksisiteye kadar hastalar�n TECENTRIQ ile tedavi edilmeleri �nerilmektedir.
Hastal�k progresyonundan sonra devam eden TECENTRIQ tedavisiyle atipik yan�tlar (yani ilk olarak hastal�k progresyonu ve ard�ndan t�m�rde k���lme) g�zlenmi�tir. Hekimin takdirine ba�l� olarak hastal�k progresyonundan sonra tedavi uygulanmas� d���n�lebilir.
Geciken veya atlanan dozlar:
Planlanm�� bir TECENTRIQ dozu atlan�rsa m�mk�n olan en k�sa s�rede uygulanmal�d�r. Uygulama plan�, dozlar aras�nda 3 haftal�k bir aral�k korunacak �ekilde ayarlanmal�d�r.
Tedavi s�ras�nda doz modifikasyonlar�:
TECENTRIQ i�in doz azalt�m� �nerilmez.
�mm�nite ile ili�kili advers reaksiyon | �iddet | Tedavi modifikasyonu |
Pn�monit | 2. derece | TECENTRIQ tedavisine ara verilir.
Olay 12 hafta i�inde 0. derece veya 1. dereceye iyile�ti�inde ve kortikosteroidler g�nde ≤10 mg prednizon e�de�erine d���r�ld���nde tedaviye devam edilebilir. |
3. veya 4. derece | TECENTRIQ tedavisi tamamen kesilir. | |
Hepatosel�ler kanseri olmayan hastalarda hepatit | 2. derece: (ALT veya AST >3-5 x normalin �st s�n�r� [N�S] veya
kan bilirubin >1,5-3 x N�S) | TECENTRIQ tedavisine ara verilir.
Olay 12 hafta i�inde 0. derece veya 1. dereceye iyile�ti�inde ve kortikosteroidler g�nde ≤10 mg prednizon veya e�de�erine d���r�ld���nde tedaviye devam edilebilir. |
3. veya 4. derece: (ALT veya AST >5 x N�S
veya
kan bilirubin >3 x N�S) | TECENTRIQ tedavisi tamamen kesilir. | |
Hepatosel�ler kanserli hastalarda hepatit | E�er AST veya ALT ba�lang��ta normal seviyelerde olup >3 x ila ≤10 x N�S'e y�kselirse
veya
E�er AST veya ALT ba�lang��ta >1 x ila ≤3 x N�S olup >5 x ila ≤10 x N�S'e y�kselirse
veya | TECENTRIQ tedavisine ara verilir.
Olay 12 hafta i�inde 0. derece veya 1. dereceye iyile�ti�inde ve kortikosteroidler g�nde ≤10 mg prednizon veya e�de�erine d���r�ld���nde tedaviye devam edilebilir. |
Doz gecikmesi veya kesilmesi (ayr�ca bkz. B�l�m 4.4 ve 4.8): Tablo 1: TECENTRIQ i�in doz modifikasyon tavsiyesi
�mm�nite ile ili�kili advers reaksiyon | �iddet | Tedavi modifikasyonu |
| E�er AST veya ALT ba�lang��ta >3 x ila ≤5 x N�S olup >8 x ila ≤10 x N�S'e y�kselirse |
|
E�er AST veya ALT >10 x N�S'e y�kselirse
veya
total bilirubin >3 x N�S'e y�kselirse | TECENTRIQ tedavisi tamamen kesilir. | |
Kolit | 2. veya 3. derece diyare (ba�lang�ca g�re ≥4 d��k�/g�n)
veya Semptomatik kolit | TECENTRIQ tedavisine ara verilir.
Olay 12 hafta i�inde 0. derece veya 1. dereceye iyile�ti�inde ve kortikosteroidler g�nde ≤10 mg prednizon veya e�de�erine d���r�ld���nde tedaviye devam edilebilir. |
4. derece diyare veya kolit (ya�am� tehdit edici; acil m�dahale endike) | TECENTRIQ tedavisi tamamen kesilir. | |
Hipotiroidizm veya hipertiroidizm | Semptomatik | TECENTRIQ tedavisine ara verilir.
Hipotiroidizm: Semptomlar tiroid replasman tedavisi ile kontrol alt�na al�nd���nda ve TSH d�zeyleri d��meye ba�lad���nda tedaviye devam edilebilir.
Hipertiroidizm: Semptomlar anti-tiroid bir t�bbi �r�n ile kontrol alt�na al�nd���nda ve tiroid fonksiyonu iyile�meye ba�lad���nda tedaviye devam edilebilir. |
Adrenal yetmezlik | Semptomatik | TECENTRIQ tedavisine ara verilir.
Semptomlar 12 hafta i�inde 0. derece veya 1. dereceye iyile�ti�inde ve kortikosteroidler g�nde ≤10 |
�mm�nite ile ili�kili advers reaksiyon | �iddet | Tedavi modifikasyonu |
|
| mg prednizon veya e�de�erine d���r�ld���nde ve hastan�n durumu replasman tedavisinde stabil hale geldi�inde tedaviye devam edilebilir. |
Hipofizit | 2. veya 3. derece | TECENTRIQ tedavisine ara verilir.
Semptomlar 12 hafta i�inde 0. derece veya 1. dereceye iyile�ti�inde ve kortikosteroidler g�nde ≤10 mg prednizon veya e�de�erine d���r�ld���nde ve hastan�n durumu replasman tedavisinde stabil hale geldi�inde tedaviye devam edilebilir. |
4. derece | TECENTRIQ tedavisi tamamen kesilir. | |
Tip 1 diabetes mellitus | 3. veya 4. derece hiperglisemi (a�l�k glukozu >250 mg/dL veya 13,9 mmol/L) | TECENTRIQ tedavisine ara verilir.
�ns�lin replasman tedavisinde metabolik kontrol elde edildi�inde tedaviye devam edilebilir. |
D�k�nt�/�iddetli kutan�z advers reaksiyonlar | 3. derece
veya ��pheli Stevens- Johnson sendromu (SJS) veya toksik epidermal nekroliz (TEN) | TECENTRIQ tedavisine ara verilir.
Semptomlar 12 hafta i�inde 0. derece veya 1. dereceye iyile�ti�inde ve kortikosteroidler g�nde ≤10 mg prednizon veya e�de�erine d���r�ld���nde tedaviye devam edilebilir. |
4. derece
veya do�rulanm�� Stevens- Johnson sendromu (SJS) veya toksik epidermal nekroliz (TEN) | TECENTRIQ tedavisi tamamen kesilir. | |
Miyastenik sendrom/ miyastenia gravis, Guillain-Barré sendromu ve meningoensefalit | T�m dereceler | TECENTRIQ tedavisi tamamen kesilir. |
�mm�nite ile ili�kili advers reaksiyon | �iddet | Tedavi modifikasyonu |
Pankreatit | Serum amilaz veya lipaz d�zeylerinde 3. veya 4. derece y�kselme (> 2 N�S) veya 2. veya 3. derece pankreatit | TECENTRIQ tedavisine ara verilir.
Serum amilaz ve lipaz d�zeyleri 12 hafta i�inde 0. derece veya 1. dereceye iyile�ti�inde veya pankreatit semptomlar� d�zeldi�inde ve kortikosteroidler g�nde ≤10 mg prednizon veya e�de�erine d���r�ld���nde TECENTRIQ ile tedaviye devam edilebilir. |
4. derece veya herhangi bir derecede n�kseden pankreatit | TECENTRIQ tedavisi tamamen kesilir. | |
Miyokardit | 2. derece veya �zeri | TECENTRIQ tedavisi tamamen kesilir. |
Nefrit | 2. derece (kreatinin d�zeyi > ba�lang�ca g�re 1,5 – 3 x veya >1,5 – 3 x N�S) | TECENTRIQ tedavisine ara verilir.
Olay 12 hafta i�inde 0. derece veya 1. dereceye iyile�ti�inde ve kortikosteroidler g�nde ≤10 mg prednizon veya e�de�erine d���r�ld���nde tedaviye devam edilebilir. |
3. veya 4. derece (kreatinin d�zeyinde ba�lang�ca g�re 3 kat art�� veya N�S'e g�re 3 kat art��) | TECENTRIQ tedavisi tamamen kesilir. | |
Miyozit | 2. veya 3. derece | TECENTRIQ tedavisine ara verilir |
4. derece veya 3. derecede n�kseden miyozit | TECENTRIQ tedavisi tamamen kesilir. | |
Perikardiyal bozukluklar | 1. derece perikardit | TECENTRIQ tedavisine ara verilir |
2. derece veya �zeri | TECENTRIQ tedavisi tamamen kesilir | |
Hemofagositik lenfohistiyositoz | ��pheli hemofagositik lenfohistiyositoz | TECENTRIQ tedavisi tamamen kesilir |
�mm�nite ile ili�kili di�er advers reaksiyonlar | 2. veya 3. derece | Olay 12 hafta i�inde 0. derece veya 1. dereceye iyile�ene ve kortikosteroidler g�nde ≤10 mg prednizon veya |
�mm�nite ile ili�kili advers reaksiyon | �iddet | Tedavi modifikasyonu |
|
| e�de�erine d���r�lene kadar TECENTRIQ tedavisine ara verilir. |
4. derece veya 3. derece n�kseden advers olaylar | TECENTRIQ tedavisi tamamen kesilir. (replasman hormonlar�yla kontrol alt�na al�nan endokrinopatiler hari�) | |
Di�er Advers reaksiyonlar | �iddet | Tedavi Modifikasyonu |
�nf�zyonla ili�kili reaksiyonlar | 1. veya 2. derece | �nf�zyon h�z� azalt�l�r veya kesilir.
Olay d�zeldikten sonra tedavi s�rd�r�lebilir. |
3. veya 4. derece | TECENTRIQ tedavisi tamamen kesilir. |
Not: Toksisite dereceleri, Ulusal Kanser Enstit�s� Advers Olaylar i�in Ortak Terminoloji Kriterleri, Versiyon 4.0'a (NCI-CTCAE v.4) uygundur.
TECENTRIQ ile tedavi edilen hastalara ilac�n riskleri hakk�nda bilgi veren Hasta Uyar� Kartlar� verilmelidir.
Uygulama �ekli:
TECENTRIQ intraven�z kullan�ma y�neliktir. TECENTRIQ inf�zyonlar� intraven�z pu�e veya bolus �eklinde uygulanmamal�d�r.
�lk TECENTRIQ dozu 60 dakika uygulanmal�d�r. �lk inf�zyon tolere edilirse, sonraki t�m inf�zyonlar 30 dakikada uygulanabilir.
T�bbi �r�n�n uygulanmadan �nceden seyreltilmesi ve kullan�m�na ili�kin talimatlar i�in B�l�m
6.6'ya bak�n�z.
�zel pop�lasyonlara ili�kin ek bilgiler:
Karaci�er yetmezli�i:
Pop�lasyon farmakokinetik analizine g�re hafif veya orta d�zeyde karaci�er bozuklu�u olan hastalarda doz ayarlamas� gerekli de�ildir. �iddetli karaci�er bozuklu�u olan hastalara ili�kin veri mevcut de�ildir (bkz. B�l�m 5.2).
B�brek yetmezli�i:
Pop�lasyon farmakokinetik analizine g�re hafif ve orta derecede b�brek bozuklu�u olan hastalarda doz ayarlamas� gerekli de�ildir (bkz. B�l�m 5.2). �iddetli b�brek bozuklu�u olanlara ait bilgi, bu pop�lasyonda bir sonuca varmak i�in �ok azd�r.
Pediyatrik pop�lasyon:
TECENTRIQ'in �ocuklarda ve 18 ya� alt�ndaki adolesanlarda g�venlili�i ve etkilili�i g�sterilmemi�tir. Mevcut olan veriler B�l�m 4.8, 5.1 ve 5.2'de anlat�lm��t�r ancak pozoloji ile ilgili herhangi bir tavsiye verilememektedir.
Geriyatrik pop�lasyon:
Pop�lasyon farmakokinetik analizine g�re 65 ya� ve �st�ndeki hastalarda TECENTRIQ doz ayarlamas� gerekli de�ildir (bkz. B�l�m 4.8 ve 5.1).
Do�u Kooperatif Onkoloji Grubu (ECOG) performans stat�s� ≥2
ECOG performans stat�s� ≥2 olan hastalar K���k H�creli D��� Akci�er Kanseri (KHDAK), Erken Evre K���k H�creli Akci�er Kanseri (EE-KHAK), 2. Basamak �rotelyal Kanser ve Hepatosel�ler Kanser klinik �al��malar�na dahil edilmemi�tir (bkz. B�l�m 4.4 ve 5.1).
4.3. Kontrendikasyonlar
TECENTRIQ'in etkin maddesi atezolizumaba veya B�l�m 6.1'de listelenen yard�mc� maddelerden herhangi birine a��r� duyarl�l��� olan hastalarda kontrendikedir.
4.4. �zel kullan�m uyar�lar� ve �nlemleri
�mm�nite ile ili�kili advers reaksiyonlar:TECENTRIQ ile tedavi s�ras�nda olu�an imm�nite ile ili�kili advers reaksiyonlar�n �o�u ilac�n kesilmesi ve kortikosteroidlerin veya destekleyici tedavinin ba�lat�lmas�yla geri d�nd�r�lebilir olmu�tur. Birden fazla v�cut sistemini etkileyen imm�nite ile ili�kili advers reaksiyonlar g�r�lm��t�r ve bu reaksiyonlar TECENTRIQ'in son dozundan sonra da olu�abilir.
�mm�nite ile ili�kili ��pheli advers reaksiyonlar i�in etyolojiyi do�rulamak veya di�er nedenleri d��lamak i�in yeterli de�erlendirme yap�lmal�d�r. Advers etkilerin �iddetine ba�l� olarak, TECENTRIQ tedavisine ara verilir ve kortikosteroid uygulan�r. Olay ≤ 1. dereceye iyile�ti�inde kortikosteroid kullan�m� ≥1 ay boyunca azalt�larak kesilmelidir. �mm�nite ile ili�kili istenmeyen reaksiyonlar�n kortikosteroid kullan�m� ile kontrol edilemedi�i hastalarda, klinik �al��malardan elde edilen s�n�rl� verilere dayanarak, di�er sistemik immunosupresan ajanlar�n kullan�m� d���n�lebilir.
Herhangi bir 3. derece imm�nite ile ili�kili advers reaksiyon ikinci defa ortaya ��karsa ve replasman hormonlar ile kontrol edilen endokrinopatiler hari� herhangi bir 4. derece imm�nite ile ili�kili advers reaksiyon g�r�l�rse TECENTRIQ tedavisi tamamen kesilmelidir (bkz. B�l�m 4.2 ve 4.8).
�mm�nite ile ili�kili pn�monit:
TECENTRIQ ile y�r�t�len klinik �al��malarda �l�mc�l vakalar da dahil olmak �zere pn�monit vakalar� g�zlenmi�tir (bkz. B�l�m 4.8). Hastalar pn�monit belirtileri ve semptomlar� i�in izlenmeli ve imm�nite ile ili�kili pn�monit d���ndaki sebepler d��lanmal�d�r.
2. derece pn�monit durumunda TECENTRIQ ile tedaviye ara verilmeli ve g�nde 1-2 mg/kg
v�cut a��rl��� prednizon veya e�de�eri ile tedavi ba�lat�lmal�d�r. Semptomlar ≤1. dereceye iyile�irse kortikosteroidler ≥1 ay boyunca azalt�larak kesilmelidir. Olay 12 hafta i�inde ≤1. dereceye iyile�irse ve kortikosteroidler g�nde ≤10 mg prednizon veya e�de�erine d���r�l�rse TECENTRIQ ile tedaviye devam edilebilir. 3. veya 4. derece pn�monit durumunda TECENTRIQ ile tedavi kal�c� olarak b�rak�lmal�d�r.
�mm�nite ile ili�kili hepatit:
TECENTRIQ ile y�r�t�len klinik �al��malarda baz�lar� �l�mc�l sonu�lara yol a�an hepatit vakalar� g�zlenmi�tir (bkz. B�l�m 4.8). Hastalar hepatit belirtileri ve semptomlar� i�in izlenmelidir.
Aspartat aminotransferaz (AST), alanin aminotransferaz (ALT) ve bilirubin, TECENTRIQ ile tedavi �ncesinde ve tedavi s�ras�nda periyodik olarak ve klinik �al��malarda belirtildi�i gibi izlenmelidir.
Hepatosel�ler kanseri olmayan hastalarda 2. derece olay (ALT veya AST >3-5 x N�S veya kan bilirubin >1,5-3 x N�S) 5-7 g�nden uzun s�re devam ederse TECENTRIQ ile tedaviye ara verilmeli ve g�nde 1-2 mg/kg v�cut a��rl��� prednizon veya e�de�eri ile tedavi ba�lat�lmal�d�r. Olaylar ≤1. dereceye iyile�irse kortikosteroidler ≥1 ay boyunca azalt�larak kesilmelidir.
Olay 12 hafta i�inde ≤1. dereceye iyile�irse ve kortikosteroidler g�nde ≤10 mg prednizon veya e�de�erine d���r�l�rse TECENTRIQ ile tedaviye devam edilebilir. 3. veya 4. derece olaylarda (ALT veya AST >5 x N�S veya kan bilirubin >3 x N�S) TECENTRIQ ile tedavi kal�c� olarak b�rak�lmal�d�r.
Hepatosel�ler kanseri olan hastalarda, ALT veya AST ba�lang��taki normal s�n�rlardan >3 ila
≤10 x N�S'e; veya ba�lang��tan >1 ila ≤3 x N�S'ten >5 ila ≤10 x N�S'e; veya ba�lang��tan
>3 ila ≤5 x N�S'ten >8 ila ≤10 x N�S'e y�kselir ve 5-7 g�nden uzun s�re devam ederse TECENTRIQ ile tedaviye ara verilmeli ve g�nde 1-2 mg/kg v�cut a��rl��� prednizon veya e�de�eri ile tedavi ba�lat�lmal�d�r. Olaylar ≤1. dereceye iyile�irse kortikosteroidler ≥1 ay boyunca azalt�larak kesilmelidir.
12 hafta i�inde olaylar ≤1. dereceye iyile�irse ve kortikosteroidler g�nde ≤10 mg veya e�de�erine azalt�l�rsa TECENTRIQ tedavisine devam edilebilir. ALT veya AST >10 x N�S veya total bilirubin >3 x N�S'e y�kselirse TECENTRIQ tedavisi tamamen kesilmelidir.
�mm�nite ile ili�kili kolit:
TECENTRIQ ile y�r�t�len klinik �al��malarda diyare veya kolit vakalar� g�zlenmi�tir (bkz. B�l�m 4.8). Hastalar kolit belirtileri ve semptomlar� i�in izlenmelidir.
2. veya 3. derece diyare (ba�lang�ca g�re ≥4 d��k�/g�n art��) veya kolit (semptomatik) durumunda TECENTRIQ ile tedaviye ara verilmelidir. 2. derece diyare veya kolit durumunda semptomlar >5 g�n devam ederse veya n�ksederse, g�nde 1-2 mg/kg v�cut a��rl��� prednizon veya e�de�eri ile tedavi ba�lat�lmal�d�r. 3. derece diyare veya kolit durumunda IV kortikosteroidlerle (1-2 mg/kg v�cut a��rl���/g�n metilprednizolon veya e�de�eri) tedavi ba�lat�lmal� ve semptomlar iyile�meye ba�lad���nda g�nde 1-2 mg/kg v�cut a��rl��� prednizon veya e�de�erine ge�ilmelidir. Semptomlar ≤1. dereceye iyile�irse kortikosteroidler ≥1 ay boyunca azalt�larak kesilmelidir. Olay 12 hafta i�inde ≤1. dereceye iyile�irse ve kortikosteroidler g�nde ≤10 mg prednizon veya e�de�erine d���r�l�rse TECENTRIQ ile
tedaviye devam edilebilir. 4. derece (ya�am� tehdit edici; acil m�dahale endike) diyare veya kolit durumunda TECENTRIQ ile tedavi kal�c� olarak b�rak�lmal�d�r. Kolit ile ili�kili potansiyel gastrointestinal perforasyon komplikasyonu dikkate al�nmal�d�r.
�mm�nite ile ili�kili endokrinopatiler:
TECENTRIQ ile y�r�t�len klinik �al��malarda hipotiroidizm, hipertiroidizm, adrenal yetmezlik, hipofizit ve diyabetik ketoasidoz dahil olmak �zere tip 1 diabetes mellitus vakalar� g�zlenmi�tir (bkz. B�l�m 4.8).
Hastalar endokrinopatilerin klinik belirtileri ve semptomlar� i�in izlenmelidir. Tiroid fonksiyonu TECENTRIQ ile tedavi �ncesinde ve tedavi s�ras�nda periyodik olarak izlenmelidir. Ba�lang��ta anormal tiroid fonksiyon testleri olan hastalar�n uygun �ekilde tedavi edilmesi d���n�lmelidir.
Tiroid fonksiyonu testleri anormal olan asemptomatik hastalar TECENTRIQ alabilir. Semptomatik hipotiroidizm durumunda TECENTRIQ ile tedaviye ara verilmeli ve tiroid hormonu replasman� gerekti�inde ba�lat�lmal�d�r. �zole hipotiroidizm kortikosteroidler kullan�lmadan replasman tedavisi ile y�netilebilir. Semptomatik hipertiroidizm durumunda TECENTRIQ ile tedaviye ara verilmeli ve bir anti-tiroid t�bbi �r�n gerekti�i gibi ba�lat�lmal�d�r. Semptomlar kontrol alt�na al�nd���nda ve tiroid fonksiyonu iyile�ti�inde TECENTRIQ ile tedaviye devam edilebilir.
Semptomatik adrenal yetmezlik durumunda TECENTRIQ ile tedaviye ara verilmeli ve intraven�z kortikosteroid (g�nde 1-2 mg/kg v�cut a��rl��� metilprednizolon veya e�de�eri) ile tedavi ba�lat�lmal�d�r. Semptomlar iyile�ti�inde g�nde 1-2 mg/kg v�cut a��rl��� prednizon veya e�de�eri ile tedavi uygulanmal�d�r. Semptomlar ≤1. dereceye iyile�irse kortikosteroidler ≥1 ay boyunca azalt�larak kesilmelidir. Olay 12 hafta i�inde ≤1. dereceye iyile�irse ve kortikosteroidler g�nde ≤10 mg prednizon veya e�de�erine d���r�l�rse ve hastan�n durumu replasman tedavisinde stabilse (gerekli ise) tedaviye devam edilebilir.
2. veya 3. derece hipofizit i�in TECENTRIQ kesilmeli ve intraven�z kortikosteroidler (1 ila 2 mg/kg v�cut a��rl���/g�n metilprednizolon veya e�de�eri) ile tedavi ba�lat�lmal� ve ihtiyaca g�re hormon replasman tedavisi ba�lat�lmal�d�r. Belirtiler d�zeldi�inde 1 ila 2 mg/kg v�cut a��rl���/g�n prednizon veya e�de�eri ile tedavi uygulanmal�d�r. Semptomlar ≤ 1. dereceye iyile�irse, kortikosteroidler ≥ 1 ay boyunca azalt�larak kesilmelidir. Olay, 12 hafta i�inde ≤ 1. dereceye iyile�ir ve kortikosteroidler g�nde ≤ 10 mg prednizona veya e�de�erine d���r�l�rse ve hasta replasman tedavisinde (e�er gerekli ise) stabil kal�rsa, tedaviye devam edilebilir. 4. derece hipofizit i�in TECENTRIQ tedavisi kesilmelidir.
Tip 1 diabetes mellitus i�in ins�lin tedavisi ba�lat�lmal�d�r. ≥3. derece hiperglisemi (a�l�k glukozu >250 mg/dL veya 13,9 mmol/L) durumunda TECENTRIQ ile tedaviye ara verilmelidir. �ns�lin replasman tedavisinde metabolik kontrol elde edilirse TECENTRIQ ile tedaviye devam edilebilir.
�mm�nite ile ili�kili meningoensefalit:
TECENTRIQ ile y�r�t�len klinik �al��malarda meningoensefalit g�zlenmi�tir (bkz. B�l�m 4.8). Hastalar menenjit veya ensefalitin klinik belirtileri ve semptomlar� i�in izlenmelidir.
Herhangi bir derece menenjit veya ensefalit durumunda TECENTRIQ ile tedavi kal�c� olarak
b�rak�lmal�d�r. �ntraven�z kortikosteroidler (g�nde 1-2 mg/kg v�cut a��rl��� metilprednizolon veya e�de�eri) ile tedavi ba�lat�lmal� ve hastan�n durumu iyile�ti�inde 1-2 mg/kg v�cut a��rl��� prednizon veya e�de�erine ge�ilmelidir.
�mm�nite ile ili�kili n�ropatiler:
TECENTRIQ alan hastalarda ya�am� tehdit edici olabilen miyastenik sendrom/miyastenia gravis veya Guillain-Barré sendromu g�zlenmi�tir. Hastalar motor ve duyusal n�ropati semptomlar� i�in izlenmelidir.
Herhangi bir derece miyastenik sendrom/miyastenia gravis veya Guillain-Barré sendromu durumunda TECENTRIQ ile tedavi kal�c� olarak b�rak�lmal�d�r. G�nde 1-2 mg/kg dozda prednizon veya e�de�eri ile sistemik kortikosteroidlerin ba�lat�lmas� d���n�lmelidir.
�mm�nite ile ili�kili pankreatit:
TECENTRIQ ile y�r�t�len klinik �al��malarda serum amilaz ve lipaz d�zeylerinde art��lar dahil olmak �zere pankreatit g�zlenmi�tir (bkz. B�l�m 4.8). Hastalar akut pankreatiti d���nd�ren belirtiler ve semptomlar i�in yak�ndan izlenmelidir.
Serum amilaz veya lipaz d�zeylerinde ≥3. derece art�� (> 2 N�S) veya 2. veya 3. derece pankreatit durumunda TECENTRIQ ile tedaviye ara verilmeli ve g�nde 1-2 mg/kg v�cut a��rl��� IV metilprednizolon veya e�de�eri ile tedavi ba�lat�lmal�d�r. Semptomlar iyile�ti�inde g�nde 1-2 mg/kg v�cut a��rl��� prednizon veya e�de�eri ile tedavi uygulanmal�d�r. Serum amilaz ve lipaz d�zeyleri 12 hafta i�inde ≤1. dereceye iyile�ti�inde veya pankreatit semptomlar� d�zeldi�inde ve kortikosteroidler g�nde ≤10 mg prednizon veya e�de�erine d���r�ld���nde TECENTRIQ ile tedaviye devam edilebilir. 4. derece veya herhangi bir derecede n�kseden pankreatit durumunda TECENTRIQ ile tedavi kal�c� olarak b�rak�lmal�d�r.
�mm�nite ile ili�kili miyokardit:
TECENTRIQ ile y�r�t�len klinik �al��malarda �l�mc�l vakalar da dahil olmak �zere miyokardit g�zlemlenmi�tir (bkz. B�l�m 4.8). Hastalar miyokarditi d���nd�ren belirtiler ve semptomlar a��s�ndan izlenmelidir. Miyokardit, miyozitin klinik bir belirtisi de olabilir ve uygun bir �ekilde tedavi edilmelidir.
Kardiyak veya kardiyopulmoner semptomlar� olan hastalar, uygun �nlemlerin erken evrede ba�lat�lmas�n� sa�lamak i�in potansiyel miyokardit a��s�ndan de�erlendirilmelidir. Miyokardit ��phesi varsa, TECENTRIQ ile tedaviye ara verilmeli ve g�nde 1-2 mg/kg v�cut a��rl��� prednizon veya e�de�eri ile sistemik kortikosteroidler ba�lat�lmal� ve g�ncel klinik k�lavuzlara g�re tan�sal incelemeler ile birlikte h�zl� kardiyoloji kons�ltasyonu yap�lmal�d�r. Miyokardit tan�s� konuldu�unda, ≥2. derece miyokardit durumunda TECENTRIQ ile tedavi kal�c� olarak b�rak�lmal�d�r (bkz. B�l�m 4.2).
�mm�nite ile ili�kili nefrit:
TECENTRIQ ile y�r�t�len klinik �al��malarda nefrit g�zlemlenmi�tir (bkz. B�l�m 4.8). Hastalar renal fonksiyonda de�i�iklikler a��s�ndan yak�ndan izlenmelidir.
2. derece nefrit durumunda TECENTRIQ ile tedaviye ara verilmeli ve g�nde 1-2 mg/kg v�cut a��rl��� prednizon veya e�de�eri ile sistemik kortikosteroidler ba�lat�lmal�d�r. Olay, 12 hafta
i�inde ≤1. dereceye iyile�ir ve kortikosteroidler g�nde ≤ 10 mg prednizona veya e�de�erine d���r�ld���nde tedaviye devam edilebilir. 3. veya 4. derece nefrit durumunda TECENTRIQ ile tedavi kal�c� olarak b�rak�lmal�d�r.
�mm�nite ile ili�kili miyozit:
TECENTRIQ ile y�r�t�len klinik �al��malarda �l�mc�l vakalar da dahil olmak �zere miyozit vakalar� g�zlenmi�tir (bkz. B�l�m 4.8). Hastalar miyozit belirtileri ve semptomlar� a��s�ndan izlenmelidir. Olas� miyoziti olan hastalar miyokardit belirtileri a��s�ndan izlenmelidir.
Bir hastada miyozit belirti ve semptomlar� geli�irse, yak�n takibe al�nmal� ve hasta vakit kaybetmeden de�erlendirme ve tedavi i�in bir uzmana y�nlendirilmelidir. 2. veya 3. derece miyozit durumunda TECENTRIQ ile tedaviye ara verilmeli ve g�nde 1-2 mg/kg v�cut a��rl��� prednizon veya e�de�eri ile sistemik kortikosteroidler ba�lat�lmal�d�r. Semptomlar ≤ 1. dereceye iyile�irse, klinik olarak belirtildi�i gibi kortikosteroidler azalt�larak kesilebilir. Olay, 12 hafta i�inde ≤1. dereceye iyile�ir ve kortikosteroidler g�nde ≤ 10 mg prednizona veya e�de�erine d���r�ld���nde tedaviye devam edilebilir. 4. derece. veya 3. derece tekrarlayan miyozit durumunda ya da kortikosteroid dozu ba�lang��tan sonraki 12 hafta i�inde g�nde ≤ 10 mg prednizon e�de�erine d���r�lemedi�inde TECENTRIQ ile tedavi kal�c� olarak b�rak�lmal�d�r.
�mm�nite ile ili�kili �iddetli kutan�z advers reaksiyonlar:
TECENTRIQ ile tedavi edilen hastalarda, Stevens-Johnson sendromu (SJS) ve toksik epidermal nekroliz (TEN) dahil olmak �zere imm�nite ile ili�kili �iddetli kutan�z advers reaksiyonlar bildirilmi�tir. Hastalar ��pheli �iddetli cilt reaksiyonlar� a��s�ndan izlenmeli ve di�er sebepler d��lanmal�d�r. ��pheli �iddetli kutan�z advers reaksiyonlar� varl���nda hastalar daha ileri te�his ve takip i�in bir uzmana y�nlendirilmelidir.
Advers reaksiyonlar�n �iddetine ba�l� olarak, 3. derece cilt reaksiyonu durumunda TECENTRIQ ile tedaviye ara verilmeli ve g�nde 1-2 mg/kg v�cut a��rl��� prednizon veya e�de�eri ile sistemik kortikosteroidler ba�lat�lmal�d�r. Olay, 12 hafta i�inde ≤1. dereceye iyile�ir ve kortikosteroidler g�nde ≤ 10 mg prednizona veya e�de�erine d���r�ld���nde TECENTRIQ tedavisine devam edilebilir. 4. derece cilt reaksiyonlar� durumunda TECENTRIQ ile tedavi kal�c� olarak b�rak�lmal� ve kortikosteroid uygulanmal�d�r.
Stevens-Johnson sendromu (SJS) ve toksik epidermal nekroliz (TEN) ��phesi olan hastalarda TECENTRIQ ile tedaviye ara verilmelidir. Do�rulanm�� Stevens-Johnson sendromu (SJS) ve toksik epidermal nekroliz (TEN) vakalar�nda TECENTRIQ ile tedavi kal�c� olarak b�rak�lmal�d�r.
Daha �nce di�er imm�n sistemi stim�le edici ajanlarla tedavi s�ras�nda �iddetli veya hayat� tehdit eden cilt advers reaksiyonlar ya�ayan hastalar�n TECENTRIQ tedavisine ba�lanmas� dikkatli olarak de�erlendirilmelidir.
�mm�nite ile ili�kili perikardiyal bozukluklar
TECENTRIQ ile perikardit, perikardiyal ef�zyon ve kardiyak tamponad gibi baz�lar� �l�mc�l sonu�lara yol a�an perikardiyal bozukluklar g�zlenmi�tir (bkz. B�l�m 4.8). Hastalar, perikardiyal bozukluklar�n klinik belirtileri ve semptomlar� a��s�ndan izlenmelidir.
1. derece perikardit ��phesi durumunda TECENTRIQ tedavisi durdurulmal� ve mevcut klinik k�lavuzlara g�re tan�sal �al��malarla derhal kardiyoloji kons�ltasyonu ba�lat�lmal�d�r. ≥ 2. derece perikardiyal bozukluklardan ��phelenildi�inde, TECENTRIQ tedavisi kesilmeli, 1 ila 2 mg/kg v�cut a��rl���/g�n prednizon veya e�de�eri dozunda sistemik kortikosteroidlerle acil tedaviye ba�lanmal� ve mevcut klinik k�lavuzlar do�rultusunda tan�sal tetkik ile derhal kardiyoloji kons�ltasyonu ba�lat�lmal�d�r. Bir perikardiyal bozukluk olay� tan�s� konulduktan sonra, ≥ 2. derece perikardiyal bozukluklar i�in TECENTRIQ tedavisi kal�c� olarak kesilmelidir (bkz. B�l�m 4.2).
Hemofagositik lenfohistiyositoz
TECENTRIQ alan hastalarda �l�mc�l vakalar dahil olmak �zere hemofagositik lenfohistiyositoz (HLH) bildirilmi�tir (bkz. B�l�m 4.8). Atipik veya uzam�� sitokin sal�n�m sendromu varl��� durumunda HLH de�erlendirilmelidir. Hastalar HLH'nin klinik belirtileri ve semptomlar� a��s�ndan izlenmelidir. ��pheli HLH durumunda TECENTRIQ kal�c� olarak kesilmeli ve hastalar ileri tan� ve tedavi i�in bir uzmana y�nlendirilmelidir.
�mm�nite ile ili�kili di�er advers reaksiyonlar:
TECENTRIQ'in etki mekanizmas� g�z �n�nde bulunduruldu�unda, enfektif olmayan sistitin de dahil oldu�u imm�nite ile ili�kili di�er potansiyel advers reaksiyonlar g�r�lebilir.
Di�er sebepleri d��lamak i�in t�m ��pheli imm�nite ile ili�kili advers reaksiyonlar� de�erlendiriniz. Hastalar imm�nite ile ili�kili advers reaksiyonlar�na ait i�aretler ve semptomlar a��s�ndan g�zlenmeli ve reaksiyonun �iddetine g�re, klinik gereklili�e g�re tedavi modifikasyonu ve kortikosteroidler ile y�netilmelidir (Bkz. B�l�m 4.2 ve 4.8).
�nf�zyon ile ili�kili reaksiyonlar:
TECENTRIQ ile inf�zyon ile ilgili reaksiyonlar g�zlenmi�tir (bkz. B�l�m 4.8).
1. veya 2. derece inf�zyon ile ili�kili reaksiyon g�r�ld���nde inf�zyon h�z� d���r�lmeli veya tedavi kesilmelidir. 3. veya 4. derece inf�zyon ile ilgili reaksiyon g�r�ld���nde TECENTRIQ tedavisi tamamen sonland�r�lmal�d�r. 1. veya 2. derece inf�zyon ile ili�kili reaksiyon g�r�len hastalar yak�ndan izlenerek TECENTRIQ almaya devam edebilir; bu hastalarda antipiretik ve antihistaminiklerle premedikasyon de�erlendirilebilir.
Hastal��a spesifik �nlemler
TECENTRIQ'in, daha �nce tedavi almam�� sisplatine uygun olmayan �rotelyal kanseri olan hastalarda kullan�lmas�
Imvigor210 Kohort 1 �al��ma pop�lasyonunun ba�lang�� ve prognostik hastal�k karakterizasyonu, klinikteki sisplatine uygun olmayan ama karboplatin bazl� kombinasyon kemoterapi kullan�m�na uygun olan hastalar ile genel olarak kar��la�t�rabilir olmu�tur. Herhangi bir kemoterapi i�in uygun olmayan hastalara ait alt grup i�in veriler yetersizdir, bu nedenle TECENTRIQ bu hastalarda dikkatli kullan�lmal� ve bireysel olarak potansiyel risk ve yarar dengesi dikkatli olarak de�erlendirilmelidir.
Hepatosel�ler kanseri olan hastalarda TECENTRIQ'in bevacizumab ile birlikte kullan�lmas�
Child-Pugh B karaci�er hastal��� olan, TECENTRIQ ile bevacizumab kombinasyon tedavisi
alan HSK hastalar�na ait veriler �ok s�n�rl�d�r ve mevcut durumda Child-Pugh C karaci�er hastal��� olan HSK hastalar�na ait veri bulunmamaktad�r.
Bevacizumab ile tedavi edilen hastalarda hemoraji riski y�ksektir ve TECENTRIQ ile birlikte bevacizumab kullanan hepatosel�ler kanserli hastalarda �l�mc�l olaylar� da kapsayan �iddetli gastrointestinal hemoraji vakalar� bildirilmi�tir. Hepatosel�ler kanseri olan hastalarda TECENTRIQ ile birlikte bevacizumab tedavisine ba�lamadan �nce klinik pratik olarak �zofageal varislerin taranmas� ve tedavisi yap�lmal�d�r. Kombinasyon tedavisi ile 3. veya 4. derece kanama ge�iren hastalarda bevacizumab tedavisi tamamen kesilmelidir. Konu ile ilgili bevacizumab K�sa �r�n Bilgisine bak�n�z.
TECENTRIQ ile birlikte bevacizumab tedavisi s�ras�nda diyabetes mellitus ortaya ��kabilir. Hekimler TECENTRIQ ile birlikte bevacizumab tedavisi �ncesinde ve periyodik olarak tedavi boyunca klinik olarak gerekliliklere g�re kan glukoz seviyelerini takip etmelidir.
1. Basamak K���k H�creli D��� Akci�er Kanseri tedavisinde monoterapi olarak TECENTRIQ kullan�m�
Hekimler K���k H�creli D��� Akci�er Kanseri (KHDAK) olan hastalarda monoterapi olarak 1. Basamak tedavisine ba�lamadan �nce TECENTRIQ'in gecikmeli etkisini de�erlendirmelidir. TECENTRIQ ile kemoterapi kar��la�t�rmas�nda randomizasyondan sonraki 2,5 ay boyunca g�r�len daha y�ksek �l�m vaka say�s�n� uzun s�reli sa�kal�m faydas� takip etmi�tir. Erken �l�m vakalar� ile ilgili spesifik fakt�r(ler) tespit edilmemi�tir (bkz. B�l�m 5.1).
Klinik �al��malardan d��lanan hastalar:
A�a��daki ko�ullara sahip hastalar klinik �al��malara dahil edilmemi�tir: Otoimm�n hastal�k ge�mi�i, pn�monit ge�mi�i, aktif beyin metastaz�, HIV, hepatit B ya da hepatit C enfeksiyonu (Hepatosel�ler kanseri olmayan hastalar i�in), �nemli kardiyovask�ler hastal��� ve yetersiz hematolojik ve hedef organ fonksiyonu. Kay�ttan 28 g�n �nce canl�, zay�flat�lm�� bir a�� uygulanan hastalar, �al��maya ba�lamadan �nceki 4 hafta i�erisinde sistemik imm�n sistemi uyar�c� ajanlar veya 2 hafta i�erisinde sistemik immunosupresif ajanlar kullanan hastalar ve �al��maya ba�lamadan 2 hafta �nce terap�tik oral veya IV antibiyotik kullanan hastalar klinik �al��malara al�nmam��t�r.
Biyoteknolojik �r�nlerin takip edilebilirli�i:
Biyoteknolojik �r�nlerin takip edilebilirli�inin sa�lanmas� i�in uygulanan �r�n�n ticari ismi ve seri numaras� mutlaka hasta dosyas�na kaydedilmelidir.
Hasta Uyar� Kart�:
TECENTRIQ re�ete eden hekimler, TECENTRIQ tedavisinin risklerini hastalar�na a��klamal�d�r. TECENTRIQ ile tedavi edilen hastalara ilac�n riskleri hakk�nda bilgi veren Hasta Uyar� Kartlar� verilmeli ve kart� her zaman yanlar�nda ta��malar� s�ylenmelidir.
4.5. Di�er t�bbi �r�nler ile etkile�imler ve di�er etkile�im �ekilleri
TECENTRIQ ile herhangi bir resmi farmakokinetik ila� etkile�imi �al��mas� yap�lmam��t�r. TECENTRIQ dola��mdan katabolizma ile temizlendi�inden metabolik ila�-ila� etkile�imleri beklenmemektedir.
TECENTRIQ ile tedaviye ba�lamadan �nce, TECENTRIQ'in farmakodinamik aktivitesine ve etkilili�ine yapabilecekleri potansiyel etkiler nedeniyle sistemik kortikosteroidlerin veya immunosupresanlar�n kullan�lmas�ndan ka��n�lmal�d�r. Bununla birlikte, sistemik kortikosteroidler veya di�er immunosupresif maddeler, TECENTRIQ tedavisine ba�lad�ktan sonra imm�nite ile ili�kili advers reaksiyonlar�n tedavisinde kullan�labilir (bkz. B�l�m 4.4).
�zel pop�lasyonlara ili�kin ek bilgiler:
TECENTRIQ ile herhangi bir farmakokinetik ila� etkile�imi �al��mas� yap�lmam��t�r.
Pediyatrik pop�lasyon:
TECENTRIQ ile pediyatrik pop�lasyonda herhangi bir farmakokinetik ila� etkile�imi �al��mas� yap�lmam��t�r.
4.6. Gebelik ve laktasyon
:Gebelik kategorisi: D
�ocuk do�urma potansiyeli bulunan kad�nlar/do�um kontrol� (kontrasepsiyon):
�ocuk do�urma potansiyeline sahip kad�nlar TECENTRIQ ile tedavi s�ras�nda ve tedaviden 5 ay sonraya kadar etkili bir do�um kontrol y�ntemi kullanmal�d�r.
Gebelik d�nemi:
TECENTRIQ'in hamile kad�nlar �zerinde etkisine dair herhangi bir veri bulunmamaktad�r. TECENTRIQ ile geli�imsel �al��malar ve �reme �al��malar� yap�lmam��t�r. Hayvan �al��malar�yla, PD-L1/PD-1 yolak inhibisyonunun fare gebelik modellerinde imm�nite ile ili�kili, fet�s �l�m�yle sonu�lanan fet�s geli�iminin reddine sebep oldu�u g�sterilmi�tir (bkz. B�l�m 5.3). Bu sonu�lar, etki mekanizmas�na ba�l� olarak potansiyel bir risk olu�turmakta olup, gebelik d�neminde TECENTRIQ uygulamas�n�n artm�� d���k veya �l� do�um oranlar� dahil olmak �zere fetal zarara sebep olabilece�ini g�stermektedir.
TECENTRIQ bir insan G1 immunoglobulinidir (IgG1) ve IgG1'in plasenta bariyerini a�t��� bilinmektedir. Bu nedenle, TECENTRIQ'in anneden geli�mekte olan fet�se ge�me potansiyeli bulunmaktad�r.
Gebe kad�nlar�n klinik durumu TECENTRIQ ile tedavi gerektirmedik�e gebelik s�ras�nda TECENTRIQ kullan�lmamal�d�r.
Laktasyon d�nemi:
TECENTRIQ'in anne s�t�ne ge�ip ge�medi�i bilinmemektedir. TECENTRIQ bir monoklonal antikordur ve ilk gelen s�tte bulunmas� ve daha sonra da az miktarda s�tte bulunmas� beklenmektedir. Yeni do�anlar ve infantlar �zerindeki risk d��lanamaz. Emzirmenin �ocuk i�in faydalar� ve tedavinin anne i�in faydalar� dikkate al�narak emzirmenin durdurulmas� veya TECENTRIQ tedavisinin durdurulmas� kararla�t�r�lmal�d�r.
�reme yetene�i/fertilite:
TECENTRIQ'in fertilite �zerindeki olas� etkilerine ili�kin veri bulunmamaktad�r. TECENTRIQ'in do�urganl�k �zerindeki etkisini de�erlendirme ama�l� reprod�ktif ve geli�imsel toksisite �al��malar� yap�lmam��t�r. Bununla birlikte, 26 haftal�k yinelenen doz toksisitesi �al��mas�nda, �nerilen dozda TECENTRIQ kullanan hastalarda menstr�el siklusta e�ri alt�nda kalan alan� yakla��k 6 kat�na ��kard��� ve geri d�n���ml� oldu�u g�r�lm��t�r (bkz. B�l�m 5.3). Erkek reprod�ktif organlar� �zerinde etki g�r�lmemi�tir.
4.7. Ara� ve makine kullan�m� �zerindeki etkiler
TECENTRIQ'in ara� ve makine kullanma yetene�i �zerinde k���k bir etki g�stermesi s�z konusudur. Yorgunluk hisseden hastalara semptomlar hafifleyene kadar ara� ve makine kullanmamalar� tavsiyesinde bulunulmal�d�r (bkz. B�l�m 4.8).
4.8. �stenmeyen etkiler
G�venlilik profilinin �zeti:TECENTRIQ monoterapisinin g�venlili�i, �oklu t�m�r tiplerinde 4349 hastadaki havuzlanm�� verilere dayand�r�lm��t�r. En yayg�n advers reaksiyonlar (>%10); yorgunluk (%30,1), i�tah kayb� (%21,3), bulant� (%20), d�k�nt� (%19,3), pireksi (%19), �ks�r�k (%18,6), diyare (%18),
dispne (%17,2), artralji (%16,7), asteni (%13,2), pir�rit (%13,2), s�rt a�r�s� (%12,8), kusma (%12,5), idrar yolu enfeksiyonu (%11,5) ve ba� a�r�s� (%10,3) olmu�tur.
TECENTRIQ'in di�er t�bbi �r�nlerle kombinasyonunda kullan�m�n�n g�venlili�i, �oklu t�m�r tiplerinde 4535 hastada de�erlendirilmi�tir. En yayg�n advers reaksiyonlar (≥%20); anemi (%36,8), n�tropeni (36,6), bulant� (%35,5), yorgunluk (%33,1), trombositopeni (%27,1), diyare
(%27,6), d�k�nt� (%27,8), alopesi (%28,1), konstipasyon (%25,8), i�tah azalmas� (%24,7), periferal n�ropati (%24,4) olmu�tur.
Adjuvan K���k H�creli D��� Akci�er Kanseri ko�ulunda TECENTRIQ kullan�m�
K���k h�creli d��� akci�er kanseri (KHDAK) hasta pop�lasyonunda adjuvan ortamda TECENTRIQ'in g�venlilik profili (IMpower010), genel olarak ileri evre ortam�nda genel havuzlanm�� monoterapi g�venlilik profili ile tutarl� olmu�tur. Bununla birlikte, ileri evre hastal��� olan havuzlanm�� monoterapi pop�lasyonunda imm�n sistemle ili�kili advers reaksiyonlar�n�n insidans� %38,4 iken, IMpower010 �al��mas�nda atezolizumab�n imm�n sistemle ili�kili advers reaksiyonlar�n�n insidans� %51,7 olmu�tur. Adjuvan ortamda imm�n sistemle ilgili yeni advers reaksiyonlar saptanmam��t�r.
Ciddi advers reaksiyonlara dair detayl� bilgiler B�l�m 4.4 �zel kullan�m uyar�lar� ve �nlemleri b�l�m�nde verilmektedir.
Advers reaksiyonlar�n tablo halinde listesi
Advers �la� Reaksiyonlar� (ADR), MedDRA sistem organ s�n�f�na (SOC) ve s�kl�k kategorilerine g�re Tablo 2'de TECENTRIQ monoterapi veya kombinasyon tedavisi i�in listelenmi�tir. Kombinasyon tedavisi kullan�lan klinik �al��malarda raporlanmasa bile, tek ba��na TECENTRIQ ile veya kemoterapiler ile ortaya ��kabilece�i bilinen advers reaksiyonlar, kombinasyon tedavisi s�ras�nda ortaya ��kabilir.
A�a��daki s�kl�k kategorileri kullan�lm��t�r:
�ok yayg�n (≥1/10), yayg�n (≥1/100 ila <1/10), yayg�n olmayan (≥1/1000 ila <1/100), seyrek (≥1/10.000 ila <1/1000), �ok seyrek (<1/10.000), bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor). Her bir s�kl�k grubunda advers reaksiyonlar azalan ciddiyet s�ras�na g�re yaz�lm��t�r.
Tablo 2: TECENTRIQ ile tedavi edilen hastalarda meydana gelen advers reaksiyonlar�n �zeti
TECENTRIQ monoterapi | TECENTRIQ kombinasyon tedavisi | |
Enfeksiyonlar ve enfestasyonlar | ||
�ok yayg�n | �drar yollar� enfeksiyonu | Akci�er enfeksiyonu |
Yayg�n |
| Sepsis |
Kan ve lenf listemi hastal�klar� | ||
�ok yayg�n |
| Anemi, trombositopeni, n�tropeni, l�kopeni |
Yayg�n | Trombositopeni | Lenfopeni |
Seyrek | Hemofagositik lenfohistiyositoz | Hemofagositik lenfohistiyositoz |
Ba����kl�k sistemi hastal�klar� | ||
Yayg�n | �nf�zyonla ilgili reaksiyon | �nf�zyonla ilgili reaksiyon |
Endokrin hastal�klar | ||
�ok yayg�n |
| Hipotiroidizm |
Yayg�n | Hipotiroidizm , Hipertiroidizm | Hipertiroidizm |
Yayg�n olmayan | Diabetes mellitus, Adrenal yetmezlik |
|
Seyrek | Hipofizit |
|
Metabolizma ve beslenme hastal�klar� | ||
�ok yayg�n | ��tah kayb� | ��tah kayb� |
Yayg�n | Hipokalemi, Hiponatremi, Hiperglisemi | Hipokalemi, Hiponatremi, Hipomagnezemi |
Sinir sistemi hastal�klar� | ||
�ok yayg�n | Ba� a�r�s� | Periferal n�ropati, Ba� a�r�s� |
Yayg�n |
| Senkop, Ba� d�nmesi |
Yayg�n olmayan | Guillain-Barré sendromu, Meningoensefalit |
|
Seyrek | Miyastenik sendrom |
|
G�z Hastal�klar� | ||
Seyrek | �veit |
|
Kardiyak hastal�klar | ||
Yayg�n | Perikardiyal bozukluklar |
|
Yayg�n olmayan |
| Perikardiyal bozukluklar |
Seyrek | Miyokardit |
|
Vask�ler hastal�klar | ||
�ok yayg�n |
| Hipertansiyon |
Yayg�n | Hipotansiyon |
|
Solunum, g���s bozukluklar� ve mediastinal hastal�klar | ||
�ok yayg�n | Dispne, �ks�r�k | Dispne, �ks�r�k, nazofarenjit |
TECENTRIQ monoterapi | TECENTRIQ kombinasyon tedavisi | |
Yayg�n | Pn�monit, Hipoksi, Nazofarenjit | Disfoni |
Gastrointestinal hastal�klar | ||
�ok yayg�n | Bulant�, Kusma, Diyare | Bulant�, Kusma, Diyare, Konstipasyon |
Yayg�n | Kolit, Kar�n a�r�s�, Disfaji, Orafaringeal a�r� | Stomatit, Disguzi |
Yayg�n olmayan | Pankreatit |
|
Hepatobiliyer hastal�klar� | ||
Yayg�n | AST art���, ALT art���, Hepatit | AST art���, ALT art��� |
Deri ve deri alt� doku hastal�klar� | ||
�ok yayg�n | D�k�nt�, Pir�rit | D�k�nt�, Pir�rit, Alopesi |
Yayg�n | Ciltte kuruluk |
|
Yayg�n olmayan | �iddetli kutan�z advers reaksiyonlar, Psoriazis | �iddetli kutan�z advers reaksiyonlar, Psoriazis, |
Seyrek | Pemfigoid | Pemfigoid |
Kas-�skelet bozukluklar�, ba� doku ve kemik hastal�klar� | ||
�ok yayg�n | Artralji, S�rt a�r�s�, | Artralji, Kas-iskelet a�r�s�, S�rt a�r�s�, |
Yayg�n | Kas-iskelet a�r�s� |
|
Yayg�n olmayan | Miyozit |
|
B�brek ve idrar hastal�klar� | ||
Yayg�n | Kanda kreatinin y�kselmesi | Protein�ri, Kanda kreatinin y�kselmesi |
Yayg�n olmayan | Nefrit |
|
Bilinmiyor | Enfektif olmayan sistit |
|
Genel bozukluklar ve uygulama b�lgesine ili�kin hastal�klar | ||
�ok yayg�n | Pireksi, Yorgunluk, Asteni | Pireksi, Yorgunluk, Asteni, Periferal �dem |
Yayg�n | Grip benzeri hastal�k, �rperme |
|
Ara�t�rmalar | ||
Yayg�n |
| Kanda alkalin fosfataz y�kselmesi |
Se�ilen advers reaksiyonlar�n a��klamas�:
A�a��daki veriler, klinik �al��malarda TECENTRIQ monoterapi ile olu�an anlaml� advers reaksiyonlarla ilgili bilgileri yans�t�r (bkz. B�l�m 5.1). TECENTRIQ kombinasyonu ile ortaya ��kan advers reaksiyonlara ait detayl� bilgiler, e�er advers reaksiyon TECENTRIQ monoterapiye g�re klinik olarak anlaml� bir farkl�l�k g�steriyorsa kar��la�t�rmal� olarak verilmi�tir. Bu advers reaksiyonlar i�in y�netim k�lavuzlar� B�l�m 4.2 ve 4.4'te a��klanmaktad�r.
�mm�nite ile ili�kili pn�monit:
Pn�monit, TECENTRIQ monoterapi alan hastalar�n %3 (130/4349) kadar�nda meydana gelmi�tir. 130 hasta i�inde iki olay �l�mc�l olmu�tur. Ba�lang�ca kadar ge�en medyan s�re 4 ay (aral�k: 3 g�n – 29,8 ay) olmu�tur. Medyan s�re 1,6 ay (aral�k: 1 (bir) g�n – 27,8+ ay; "+" sans�rlenmi� bir de�eri g�sterir) olmu�tur. Pn�monit, 29 hastada (%0,7) TECENTRIQ'in b�rak�lmas�na yol a�m��t�r. Kortikosteroid kullan�m� gerektiren pn�monit, TECENTRIQ monoterapi alan hastalar�n %1,7'sinde (76/4349) meydana gelmi�tir.
�mm�nite ile ili�kili hepatit:
Hepatit, TECENTRIQ monoterapi alan hastalar�n %1,7'sinde (75/4349) meydana gelmi�tir. 75 hasta i�inde iki �l�mc�l olay olmu�tur. Ba�lang�ca kadar ge�en medyan s�re 1,6 ay (aral�k: 7 g�n – 18,8 ay) olmu�tur. Medyan s�re 2,1 ay (aral�k: 1 (bir) g�n – 22+ ay; "+" sans�rlenmi� bir de�eri g�sterir) olmu�tur. Hepatit, 13 hastada (<%0,3) TECENTRIQ'in b�rak�lmas�na yol a�m��t�r. Kortikosteroid kullan�m� gerektiren hepatit, TECENTRIQ monoterapi alan hastalar�n %0,5'inde (22/4349) meydana gelmi�tir.
�mm�nite ile ili�kili kolit:
Kolit, TECENTRIQ monoterapi alan hastalar�n %1,1'inde (50/4349) meydana gelmi�tir. Ba�lang�ca kadar ge�en medyan s�re 5,1 ay (aral�k: 15 g�n – 17,2 ay) olmu�tur. Medyan s�re 1,2 ay (aral�k: 1 (bir) g�n – 35,9+ ay; "+" sans�rlenmi� bir de�eri g�sterir) olmu�tur. Kolit, 17 hastada (%0,4) TECENTRIQ'in b�rak�lmas�na yol a�m��t�r. Kortikosteroid kullan�m� gerektiren kolit TECENTRIQ monoterapi alan hastalar�n %0,6's�nda (24/4349) meydana gelmi�tir.
�mm�nite ile ili�kili endokrinopatiler:
Tiroid bozukluklar�
Hipotiroidizm, TECENTRIQ monoterapi alan hastalar�n %7,6's�nda (331/4349) meydana gelmi�tir. Ba�lang�ca kadar ge�en medyan s�re 4,3 ay (aral�k: 1 (bir) g�n – 34,5 ay) olmu�tur. Adjuvan k���k h�creli d��� akci�er kanseri ortam�nda TECENTRIQ monoterapisi alan hastalar�n %17,4'�nde (86/495) hipotiroidizm meydana gelmi�tir. Ba�lang�ca kadar ge�en medyan s�re 4 ay (aral�k: 22 g�n - 11,8 ay) olmu�tur.
Hipertiroidizm, TECENTRIQ monoterapi alan hastalar�n %2,1'inde (93/4349) meydana gelmi�tir. Ba�lang�ca kadar ge�en medyan s�re 2,6 ay (aral�k: 1 (bir) g�n – 24,3 ay) olmu�tur. Adjuvan k���k h�creli d��� akci�er kanseri ortam�nda TECENTRIQ monoterapi alan hastalar�n %6,5'inde (32/495) hipertiroidizm meydana gelmi�tir. Ba�lang�ca kadar ge�en medyan s�re 2,8 ay (aral�k: 1 (bir) g�n - 9,9 ay) olmu�tur.
Adrenal yetmezlik
Adrenal yetmezlik, TECENTRIQ monoterapi alan hastalar�n %0,5'inde (21/4349) meydana gelmi�tir. Ba�lang�ca kadar ge�en medyan s�re 6,1 ay (aral�k: 2 (iki) g�n – 21,4 ay) olmu�tur. Adrenal yetmezlik 5 (be�) hastada (%0,1) TECENTRIQ'in b�rak�lmas�na neden olmu�tur. Kortikosteroid kullan�m� gerektiren adrenal yetmezlik TECENTRIQ monoterapi alan hastalar�n %0,4'�nde (17/4349) meydana gelmi�tir.
Hipofizit
Hipofizit, TECENTRIQ monoterapi alan hastalar�n %0,1'inden az�nda (4/4349) meydana gelmi�tir. Ba�lang�ca kadar ge�en medyan s�re 56,1 ayd�r (aral�k: 23 g�n – 13,7 ay). �� hastada (< %0,1) kortikosteroid kullan�m� gerekmi�tir ve 1 hastada (< %0,1) TECENTRIQ tedavisi durdurulmu�tur.
Hipofizit, nab-paklitaksel ve karboplatinle kombinasyon halinde atezolizumab alm�� olan hastalar�n %0,4'�nde (2/473) meydana gelmi�tir. Ba�lang�ca kadar ge�en medyan s�re, 5,2 ay (aral�k: 5,1 – 5,3 ay) olmu�tur. Her iki hastada kortikosteroid kullan�lmas� gerekmi�tir.
Diabetes mellitus
Diabetes mellitus, TECENTRIQ monoterapi alan hastalar�n % 0,5'inde (20/4349) meydana gelmi�tir. Medyan s�re 5,5 ay (aral�k: 4 (d�rt) g�n – 29 ay) olmu�tur. Diabetes mellitus, hastalar�n %0,1'inden az�nda (3/4349) TECENTRIQ'in b�rak�lmas�na yol a�m��t�r.
Diabetes mellitus, TECENTRIQ ile birlikte bevacizumab alan hepatosel�ler kanserli hastalar�n % 2,0'�nda (10/493) meydana gelmi�tir. Ba�lang�ca kadar ge�en medyan s�re 4,4 ay (aral�k: 1,2 ay – 8,3 ay) olmu�tur. TECENTRIQ tedavisinin b�rak�lmas�na neden olacak bir diabetes mellitus olay� g�r�lmemi�tir.
�mm�nite ile ili�kili meningoensefalit:
Meningoensefalit, TECENTRIQ monoterapi alan hastalar�n %0,4'�nde (18/4349) meydana gelmi�tir. Ba�lang�ca kadar ge�en medyan s�re 16 g�n (aral�k: 1 (bir) g�n – 12,5 ay) olmu�tur. Medyan s�re 22 g�n (aral�k: 6 g�n 14,5+ ay; "+" sans�rlenmi� bir de�eri g�sterir) olmu�tur.
Kortikosteroid kullan�m�n� gerektiren meningoensefalit TECENTRIQ tedavisi alan hastalar�n %0,2'sinde (10/4349) meydana gelmi�tir ve sekiz hastada (%0,2) TECENTRIQ'in b�rak�lmas�na yol a�m��t�r.
�mm�nite ile ili�kili n�ropatiler:
Guillain-Barré sendromu ve demiyalizan polin�ropati, TECENTRIQ monoterapi alan hastalar�n %0,1'inde (6/4349) meydana gelmi�tir: Bu olay i�in ba�lang�ca kadar ge�en medyan s�re 4,1 ay (aral�k: 17 g�n – 8,1 ay) olmu�tur. Medyan s�re 8 ay (aral�k: 19 g�n – 24,5 ay+, "+" sans�rlenmi� bir de�eri g�sterir). 1 (bir) hasta (< %0,1), Guillain-Barré sendromu nedeniyle TECENTRIQ kullan�m�n� b�rakm��t�r. Kortikosteroid kullan�m� gerektiren Guillain-Barré sendromu TECENTRIQ monoterapi alan hastalar�n %0,1'inden az�nda (3/4349) meydana gelmi�tir.
Miyastenik sendrom:
Miyestenia gravis, TECENTRIQ monoterapi alan hastalar�n %0,1'inden az�nda (1 (bir)/4349) meydana gelmi�tir. Ba�lang�ca kadar ge�en s�re 1,2 ayd�r.
�mm�nite ile ili�kili pankreatit:
Y�ksek amilaz ve y�ksek lipaz dahil olmak �zere pankreatit, TECENTRIQ monoterapi alan hastalar�n %0,7'sinde (32/4349) meydana gelmi�tir. Ba�lang�ca kadar ge�en medyan s�re 5,5 ay (aral�k: 1 (bir) g�n – 24,8 ay) olmu�tur. Medyan s�re 24 g�n (aral�k: 3 g�n – 22,4+ ay; "+" sans�rlenmi� bir de�eri g�sterir) olmu�tur. Pankreatit, 3 hastada (<%0,1) TECENTRIQ'in b�rak�lmas�na yol a�m��t�r. Kortikosteroid kullan�m� gerektiren pankreatit vakalar� TECENTRIQ monoterapi alan hastalar�n %0,1'inde (5/4349) meydana gelmi�tir.
�mm�nite ile ili�kili miyokardit:
Miyokardit, TECENTRIQ monoterapi alan hastalar�n %0,1'inden az�nda (3/4349) meydana gelmi�tir.Adjuvan k���k h�creli d��� akci�er kanseri ortam�nda 3 vakadan biri �l�mc�l olmu�tur. Ba�lang�ca kadar ge�en medyan s�re 2,1 ay (aral�k: 1,5 – 4,9 ay) olmu�tur. Ge�en medyan s�re 14 g�n (aral�k: 14 g�n – 2,8 ay) olmu�tur. Miyokartidiki hastada (<%0,1) TECENTRIQ'in b�rak�lmas�na neden olmu�tur. �ki hastada (< %0,1) kortikosteroid kullan�m�na gerek duyulmu�tur.
�mm�nite ile ili�kili nefrit:
Nefrit, TECENTRIQ alan hastalar�n %0,2'sinden az hastada (10/4349) meydana gelmi�tir. Ba�lang�ca kadar ge�en medyan s�re 5 ay (aral�k: 2 g�n – 17,5 ay) olmu�tur. Nefrit 5 (be�) hastada (< %0,1) TECENTRIQ'in b�rak�lmas�na neden olmu�tur. D�rt hastada (< %0,1) kortikosteroid kullan�m�na gerek duyulmu�tur.
�mm�nite ile ili�kili miyozit:
Miyozit, TECENTRIQ monoterapi alan hastalar�n %0,5'inde (20/4349) meydana gelmi�tir. Ba�lang�ca kadar ge�en medyan s�re 3,3 ayd�r (aral�k: 12 g�n ile 11 ay). Medyan s�re 5,7 ay (aral�k: 2 (iki) g�n ile 36,9 +ay; + sans�rlenmi� bir de�eri g�sterir). Miyozit 2 (iki) hastada (< %0,1) TECENTRIQ'in b�rak�lmas�na neden olmu�tur. 7 (yedi) hastada (%0,2)
kortikosteroid kullan�m�na gerek duyulmu�tur.
�mm�nite ile ili�kili �iddetli kutan�z advers reaksiyonlar:
TECENTRIQ monoterapi alan hastalar�n %0,6's�nda (28/4349) �iddetli kutan�z advers reaksiyonlar meydana gelmi�tir. 28 hastadan birinde �l�mc�l olay meydana gelmi�tir. Ba�lang�ca kadar ge�en medyan s�re 5,2 ayd�r (aral�k: 4 (d�rt) g�n ile 15,5 ay). Medyan s�re 2,4 ay (aral�k: 1 (bir) g�n ile 37,5 +ay; + sans�rlenmi� bir de�eri g�sterir). �iddetli kutan�z advers reaksiyonlar 3 (��) hastada (< %0,1) TECENTRIQ tedavisinin b�rak�lmas�na neden olmu�tur.TECENTRIQ monoterapi alan hastalar�n %0,2'sinde (9/4349) kortikosteroid kullan�m�na gerek duyulmu�tur.
�mm�nite ile ili�kili perikardiyal bozukluklar:
TECENTRIQ monoterapisi alan hastalar�n %1,1'inde (47/4349) perikardiyal bozukluklar meydana gelmi�tir. Medyan ba�lang�� s�resi 1,4 ayd�r (aral�k: 6 g�n ila 17,5 ay). Medyan s�re 1,4 ayd�r (aral�k: 0 ila 19,3+ ay; "+" sans�rlenmi� bir de�eri g�sterir). Perikardiyal bozukluklar 3 (< %0,1) hastada TECENTRIQ tedavisinin kesilmesine neden olmu�tur. Kortikosteroid kullan�m�n� gerektiren perikardiyal bozukluklar hastalar�n %0,2'sinde (7/4349) meydana gelmi�tir.
�mm�nojenisite:
�oklu faz III �al��malar� kar��la�t�r�lmas�nda, hastalar�n %13,1 ile %54,1'i tedaviye yeni ba�layan anti-ila� antikorlar� (ADA) geli�tirmi�tir. Tedavi sonucu olu�mu� anti-ila� antikoru (ADA) geli�en hastalar�n ba�lang��ta genel olarak sa�l�k durumu ve hastal�k karakteristikleri a��s�ndan daha zay�f oldu�u g�r�lm��t�r. Ba�lang��taki bu sa�l�k ve hastal�k karakteristiklerindeki dengesizlikler, farmakokinetik, etkililik ve g�venlilik analizlerinin yorumlanmas�nda kar���kl�k yaratabilmektedir. Anti-ila� antikorlar�n�n (ADA) etkilili�e etkisini ara�t�rmak i�in ba�lang��taki sa�l�k ve hastal�k karakteristiklerindeki dengesizlikleri ayarlayan ke�if analizleri yap�lm��t�r. Bu analizlerde ADA geli�tiren hastalar�n, ADA geli�tirmeyen hastalara k�yasla etkililik faydas�nda azalma ihtimali g�zard� edilmemi�tir. Anti- ila� antikorlar�n�n ba�lang�ca kadar ge�en medyan s�resi 3 (��) ila 5 (be�) hafta olmu�tur.
TECENTRIQ monoterapisi (N=3460) ve kombinasyon tedavileri (N=2285) ile tedavi edilen hastalardan elde edilen hasta havuzu verilerinden, ADA-pozitif pop�lasyonunda ADA-negatif pop�lasyonuna k�yasla g�zlenen advers olaylar�n s�kl��� s�ras�yla: Monoterapi i�in; 3. ve 4. derece advers olaylar %46,2'ye kar�� %39,4, ciddi advers olaylar %39,6'ya kar�� %33,3, tedavinin kesilmesine neden olan advers olaylar %8,5'e kar�� %7,8 iken; Kombinasyon tedavisi i�in 3. ve 4. derece advers olaylar %63,9'a kar�� %60,9, ciddi advers olaylar %43,9'a kar�� 35,6, tedavinin kesilmesine neden olan advers olaylar % 22,8'e kar�� %18,4 olmu�tur (kombinasyon tedavisi i�in). Ancak mevcut verilerden yola ��karak olas� ila� advers reaksiyonlar�n�n yola�� hakk�nda kesin sonu�lara var�lamamaktad�r.
Pediyatrik pop�lasyon
TECENTRIQ'in �ocuklar ve ad�lesanlardaki g�venlili�i bilinmemektedir. 69 pediyatrik hastada (<18 ya�) yap�lan bir klinik �al��mada yeni bir g�venlilik sinyali olu�mam��t�r ve g�venlilik profili eri�kinlerinki ile kar��la�t�r�labilirdir.
Geriyatrik pop�lasyon
TECENTRIQ monoterapi alan 65 ya� ve �zerindeki hastalar ile daha gen� hastalar aras�nda genel olarak bir g�venlilik farkl�l��� g�zlemlenmemi�tir.
Impower150, IMpower133 ve IMpower110 �al��malar�ndan elde edilen veriler, 75 ya� ve �zeri hasta grubu hakk�nda de�erlendirme yap�lmas� i�in �ok s�n�rl�d�r.
��pheli advers reaksiyonlar�n raporlanmas�
Ruhsatland�rma sonras� ��pheli ila� advers reaksiyonlar�n�n raporlanmas� b�y�k �nem ta��maktad�r. Raporlama yap�lmas�, ilac�n yarar/risk dengesinin s�rekli olarak izlenmesine olanak sa�lar. Sa�l�k mesle�i mensuplar�n�n herhangi bir ��pheli advers reaksiyonu T�rkiye Farmakovijilans Merkezi (T�FAM)'ne bildirmeleri gerekmektedir (www.titck.gov.tr; e-posta: tufam@titck.gov.tr; tel: 0 800 314 00 08; faks:0 312 218 35 99).
4.9. Doz a��m� ve tedavisi
TECENTRIQ doz a��m�na ili�kin bilgi mevcut de�ildir.
Doz a��m� durumunda, hastalar advers reaksiyon belirtileri veya semptomlar� bak�m�ndan yak�ndan izlenmeli ve uygun semptomatik tedavi ba�lat�lmal�d�r.
5. FARMAKOLOJ�K �ZELL�KLER
5.1. Farmakodinamik �zellikler
Farmakoterap�tik grup: Antineoplastikler ve imm�nomod�lat�r ajanlar, monoklonal antikorlar ve antikor ila� konjugatlar�, PD-1/PDL-1 (Programlanm�� H�cre �l�m Proteini 1/ �l�m Ligand� 1) �nhibit�rleri
ATC kodu: L01FF05
Etki mekanizmas�:
PD-L1, t�m�r h�creleri ve/veya t�m�r infiltre eden imm�n h�crelerinde eksprese olabilir ve t�m�r mikroortam�nda anti-t�m�r imm�n yan�t�n�n inhibisyonuna katk�da bulunabilir. PD- L1'in T h�crelerinde ve antijen sunan h�crelerde bulunan PD-1 ve B7.1 resept�rlerine ba�lanmas� sitotoksik T h�cresi aktivitesini, T h�cresi �o�almas�n� ve sitokin �retimini bask�lar.
Atezolizumab Fc b�lgesi de�i�tirilmi�, h�manize bir imm�noglob�lin G1 (IgG1) monoklonal bir antikorudur; do�rudan PD-L1'e ba�lan�r ve PD-1 ve B7.1 resept�rlerinin ikili blokaj�n� sa�layarak, antikor ba��ml� sell�ler sitotoksisiteyi ind�klemeden antit�m�r imm�n yan�t�n yeniden aktive edilmesi de dahil, imm�n yan�t�n PD-L1/PD-1 arac�l� inhibisyonunu serbest b�rak�r. Atezolizumab, PD-L2/PD-1 etkile�imini koruyarak PD-L2/PD-1 arac�l� inhibit�r sinyallerin devam etmesine izin verir.
Klinik etkililik ve g�venlilik:
K���k h�creli d��� akci�er kanseri:
Erken evre k���k h�creli d��� akci�er kanserinin adjuvan tedavisi
IMpower010 (GO29527): Rezeke edilmi� KHDAK'l� hastalarla sisplatin bazl� kemoterapiden sonra randomize faz III �al��ma
Evre IB (t�m�rler ≥ 4cm) – IIIA KHDAK'l� hastalar�n adjuvan tedavisi i�in atezolizumab�n etkilili�ini ve g�venlili�ini de�erlendirmek �zere bir faz III, a��k etiketli, �ok merkezli, randomize �al��ma olan GO29527 (IMpower010) y�r�t�lm��t�r (Kanser evrelendirme sistemine ili�kin Uluslararas� Kanser Kontrol� Birli�i/Amerikan Ortak Kanser Komitesi, 7. Bask�s�na g�re).
A�a��daki se�im kriterleri, terap�tik endikasyona dahil olan ve 7. bask� evreleme sistemine g�re evre II - IIIA hasta pop�lasyonunu yans�tan y�ksek n�ks riski olan hastalar� tan�mlar:
T�m�r boyutu ≥ 5 cm veya N1 veya N2 durumunun e�lik etti�i herhangi bir boyuttaki t�m�rler veya torasik yap�lara invaze olan t�m�rler (do�rudan parietal plevra, g���s duvar�, diyafram, frenik sinir, mediastinal plevra, parietal perikard, mediasten, kalp, b�y�k damarlar, trakea, rek�rren laringeal sinir, �zofagus, vertebral g�vde, karinaya invaze) veya ana bron�u karinan�n
< 2 cm distalinde tutan ancak karina tutulumu olmayan t�m�rler veya t�m akci�erin atelektazisi veya obstr�ktif pn�monisi ile ba�lant�l� t�m�rler veya primer olarak ayn� lobda veya farkl� ipsilateral lobda ayr� nod�l(ler)e sahip t�m�rler.
�al��ma, mediasten, kalp, b�y�k damarlar, trakea, rek�rren laringeal sinir, �zofagus, vertebral cisim ve karinaya invaze t�m�rleri olan N2 durumundaki hastalar� veya farkl� bir ipsilateral lobda ayr� t�m�r nod�lleri olan hastalar� i�ermemi�tir.
Toplamda 1280 kaydedilmi� hasta tam t�m�r rezeksiyonu yapt�rm�� ve 4 sikluse kadar sisplatin bazl� kemoterapi almaya uygun olmu�tur. Sisplatin bazl� kemoterapi rejimleri Tablo 3'te tarif edilmektedir.
Tablo 3: Adjuvan tedavi rejimleri (IMpower010)
Sisplatin bazl� adjuvan kemoterapi: Belirtilen tedavi rejimlerinden biri ile her 21 g�nl�k siklusun 1.g�n�nde IV olarak 75 mg/m sisplatin | 1. ve 8. g�n intraven�z olarak 30 mg/m vinorelbin |
1. g�n intraven�z olarak 75 mg/m dosetaksel | |
1. ve 8. g�n intraven�z olarak 1250 mg/m gemsitabin | |
1. g�n intraven�z olarak 500 mg/m pemetreksed (non-skuam�z) |
Sisplatin bazl� kemoterapinin (d�rt siklusa kadar) tamamlanmas�ndan sonra toplamda 1005 hasta 1:1 oran�nda atezolizumab (Kol A) veya en iyi destekleyici bak�m� (BSC) (Kol B) almaya randomize edilmi�tir. Atezolizumab, hastal�k n�ks� veya kabul edilemez toksisite g�r�lmedi�i s�rece 16 d�ng�ye kadar her 3 haftada bir IV inf�zyonla 1200 mg'l�k sabit dozda uygulanm��t�r. Randomizasyon cinsiyet, hastal�k evresi ve PD-L1 ekspresyonuna g�re s�n�fland�r�lm��t�r.
Hastalar, otoimm�n hastal�k �yk�s�ne sahiplerse; randomizasyondan �nceki 28 g�n i�inde canl�, atten�e a�� uygulamas� yap�lm��sa; randomizasyondan �nceki 4 hafta i�inde sistemik immunostimulat�r ajanlar veya 2 hafta i�inde immunosupresif ila�lar uygulanm��sa hari�
tutulmu�tur. T�m�r de�erlendirmeleri randomizasyon faz�n�n ba�lang�c�nda ve 1. siklus, 1. g�n�n�n ard�ndan ilk y�l boyunca her 4 ayda bir ve ard�ndan be� y�la kadar her 6 ayda bir ve sonras�nda y�lda bir y�r�t�lm��t�r.
ITT pop�lasyonunun demografik bilgileri ve ba�lang��taki hastal�k �zellikleri tedavi kollar� aras�nda iyi dengelenmi�tir. Medyan ya� 62 olup (aral�k: 26 ila 84) hastalar�n %67'si erkektir. Hastalar�n �o�u Beyaz (%73) ve %24'� Asyal�d�r. �o�u hasta sigara i�icisidir veya ge�mi�te i�mi�tir (%78) ve hastalarda ba�lang�� ECOG performans durumu 0 (%55) veya 1'dir (%44). Genelde hastalar�n %12'sinde evre IB, %47'sinde evre II ve %41'inde evre IIIA hastal�k mevcuttur. T�m�rleri VENTANA PD-L1 (SP263) Analizi ile �l��ld��� �zere TC'de PD-L1 ekspresyonu ≥ %1 ve ≥ %50 olan t�m�rleri olan hastalar�n y�zdesi s�ras�yla %55 ve %26 olmu�tur.
Birincil etkililik sonucu �l��m� ara�t�rmac� taraf�ndan de�erlendirilen hastal�ks�z sa�kal�md�r (HS). Hastal�ks�z sa�kal�m, randomizasyon tarihinden a�a��dakilerden birinin meydana geldi�i tarihe kadar ge�en s�re olarak tan�mlanm��t�r: ilk belgelenmi� hastal�k n�ks�, yeni primer KHDAK veya herhangi bir nedenle �l�m (hangisi daha �nce olursa). Birincil etkililik hedefi, PD-L1 ≥ %1 TC evre II IIIA hasta pop�lasyonunda HS'yi de�erlendirmektir. Kritik ikincil etkililik hedefleri, PD-L1 ≥ %50 TC evre II-IIIA hasta pop�lasyonunda HS'yi ve ITT pop�lasyonunda genel sa�kal�m� (GS) de�erlendirmektir.
Ara hastal�ks�z sa�kal�m analizi zaman�nda, �al��ma birincil sonlan�m noktas�n� sa�lam��t�r. Medyan takip s�resi yakla��k 32 ayd�r. EGFR mutasyonlar� veya ALK yeniden d�zenlemeleri olmayan PD-L1 ≥ %50 TC evre II- IIIA hastalar�n�n analizinde (n = 209), BSC koluna k�yasla atezolizumab kolunda HS'de klinik olarak anlaml� bir iyile�me g�zlenmi�tir (Tablo 4). Genel sa�kal�m (GS) verileri EGFR mutasyonlar� ve ALK yeniden d�zenlemeleri olmaks�z�n, PD-L1
≥ %50 TC evre II- IIIA hasta pop�lasyonunda genel olarak bildirilen yakla��k %16,3 oran�nda �l�m ile hastal�ks�z sa�kal�mda ara analizi s�ras�nda olgunla�mam��t�r. Genel sa�kal�m�n ke�ifsel analizi bu hasta pop�lasyonunda 0,39'luk s�n�fland�r�lm�� bir HR (%95 GA:0,18, 0,82) ile en iyi destekleyici bak�ma kar�� atezolizumab lehine bir e�ilimi d���nd�rm��t�r.
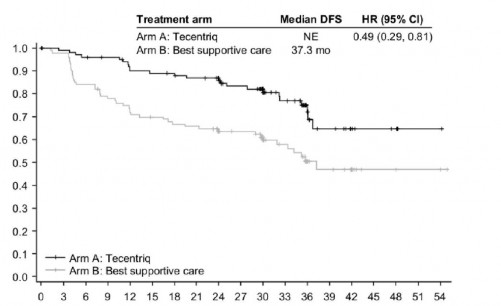
EGFR mutasyonlar� ve ALK yeniden d�zenlemeleri olmayan PD-L1 ≥ %50 TC evre II- IIIA hasta pop�lasyonu i�in ana etkililik bulgular� Tablo 4'te �zetlenmektedir. Hastal�ks�z sa�kal�m i�in Kaplan-Meier e�risi �ekil 1'de sunulmaktad�r.
Tablo 4: EGFR mutasyonlar� veya ALK yeniden d�zenlemeleri olmayan PD-L1 ekspresyonu ≥ %50 TC evre II- IIIA hasta pop�lasyonunda etkililik �zeti (IMpower010)
Etkililik sonlan�m noktas� | A kolu (Atezolizumab) | B kolu (En iyi destekleyici tedavi) |
Ara�t�rmac� taraf�ndan de�erlendirilen hastal�ks�z sa�kal�m (HS) |
n=106 |
n=103 |
Olaylar�n say�s� (%) | 24 (%22,6) | 45 (%437) |
Hastal�ks�z sa�kal�m�n (HS) medyan s�resi (ay) | NE | 37,3 |
%95 GA | NE, NE | 30,1, NE |
S�n�fland�r�lm�� risk oran� (%95 GA) | 0,49 (0,29,0,81) | |
3 y�ll�k hastal�ks�z | 75,1 | 50,4 |
sa�kal�m oran� (%)
HS=Hastal�ks�z sa�kal�m, GA=G�ven Aral���, NE=�ng�r�lemiyor
�ekil 1: EGFR mutasyonlar� veya ALK yeniden d�zenlemeleri olmayan PD-L1 ekspresyonu ≥ %50 TC evre II- IIIA hasta pop�lasyonunda (IMpower010) hastal�ks�z sa�kal�m i�in Kaplan-Meier e�risi
|
S�re (ay)
![]()
NE=�ng�r�lemiyor
En iyi destekleyici tedavi koluna k�yasla atezolizumab kolunda g�zlenen hastal�ks�z sa�kal�m iyile�mesi, hem non-skuamoz KHDAK hastalar� (s�n�fland�r�lmam�� risk oran�: 0,35 , %95 GA: 0,18, 0,69; medyan hastal�ks�z sa�kal�m NE'ye (NE= �ng�r�lemiyor) kar�� 35,7 ay) hem de skuamoz KHDAK hastalar� (s�n�fland�r�lmam�� risk oran�: 0,60 ,%95 GA: 0,29 , 1,26; medyan hastal�ks�z sa�kal�m 36,7'ye kar�� NE (NE=�ng�r�lemiyor) ay) i�eren EGFR mutasyonlar� veya ALK yeniden d�zenlemeleri olmayan PD-L1 ≥%50 TC evre II-IIIA hasta pop�lasyonunda �nceden belirtilmi� alt gruplar�n �o�u aras�nda tutarl� �ekilde g�sterilmi�tir.
Birinci basamak k���k h�creli d��� akci�er kanseri
IMpower130 (GO29537): Daha �nce kemoterapi uygulanmam�� metastatik non- skuamoz KHDAK hastalar�nda nab-paklitaksel ve karboplatinle kombinasyona y�nelik randomize faz III �al��ma.
Daha �nce kemoterapi uygulanmam�� metastatik non-skuamoz KHDAK hastalar�nda nab- paklitaksel ve karboplatinle kombinasyon halinde atezolizumab�n etkililik ve g�venlili�ini de�erlendirmek �zere bir faz III, a��k etiketli, randomize �al��ma (GO29537 (IMpower130)) ger�ekle�tirilmi�tir. EGFR mutasyonlar� veya ALK rearanjmanlar� olan hastalarda daha �nce tirozin kinaz inhibit�rleriyle tedavi alm�� olma ko�ulu aranm��t�r.
Hastalar, Amerikan Ortak Kanser Komitesi (AJCC) 7. bask�ya g�re evrelendirilmi�tir. Otoimm�n hastal�k �yk�s� olan, randomizasyondan �nceki 28 g�n i�inde canl� ve zay�flat�lm��
a�� uygulanm��, randomizasyondan �nceki 4 hafta i�inde imm�nostim�lat�r ajan veya randomizasyondan �nceki 2 hafta i�inde sistemik imm�nos�presif ila� uygulanm�� ve aktif veya tedavi edilmemi� MSS metastazlar� olan hastalar �al��maya al�nmam��t�r. Daha �nce CD137 agonistleri veya imm�n kontrol noktas� blokaj tedavileri (anti-PD-1 ve anti-PD-L1 terap�tik antikorlar�) alm�� hastalar �al��maya uygun de�ildir. Bununla birlikte, daha �nce anti- CTLA-4 tedavisi alm�� olan hastalar, en son dozun randomizasyondan en az 6 hafta �nce al�nm�� olmas� ve anti-CTLA-4'ten kaynaklanan �iddetli imm�n ili�kili advers etki �yk�s� (NCI 3 ve 4. Derece) olmamas� ko�uluyla �al��maya al�nm��t�r. T�m�r de�erlendirmeleri, 1. Siklustan sonra ilk 48 hafta boyunca 6 haftada bir ve daha sonra 9 haftada bir yap�lm��t�r. T�m�r numuneleri, t�m�r h�creleri (TC) ve t�m�r� infiltre eden imm�n h�creler (IC) �zerindeki PD-L1 ekspresyonu bak�m�ndan de�erlendirilmi�tir ve bulgular, a�a��da a��klanan analizler i�in PD-L1 ekspresyonu alt gruplar�n�n tan�mlanmas�nda kullan�lm��t�r.
EGFR mutasyonlar� veya ALK rearanjmanlar� olanlar dahil olmak �zere hastalar, Tablo 5'te a��klanan tedavi rejimlerinden biri uygulanmak �zere �al��maya al�nm�� ve 2:1 oran�nda randomize edilmi�tir. Randomizasyon gruplar�; cinsiyet, karaci�er metastazlar� varl��� ve TC ve IC �zerindeki PD-L1 ekspresyonuna g�re olu�turulmu�tur. Tedavi rejimi B alan hastalar�n hastal�k progresyonundan sonra �apraz ge�i� yaparak atezolizumab monoterapisi almas�na izin verilmi�tir.
Tablo 5: �ntraven�z tedavi rejimleri (IMpower130)
Tedavi Rejimi | �nd�ksiyon (D�rt veya alt� kez 21 g�nl�k sikluslar) | �dame (21 g�nl�k sikluslar) |
A | Atezolizumab (1200 mg) + nab-paklitaksel (100 mg/m) + karboplatin (EAA 6) | Atezolizumab (1200 mg) |
B | Nab-paklitaksel (100 mg/m) + karboplatin (EAA 6) | En iyi destekleyici tedavi veya pemetrexed |
ITT-WT olarak tan�mlanan �al��ma pop�lasyonunun (n=679) demografik �zellikleri ve ba�lang��taki hastal�k �zelliklerinin tedavi kollar� aras�nda dengeli oldu�u g�r�lm��t�r. Medyan ya� 64't�r (aral�k: 18-86 ya�). Hastalar�n �o�u erkek (%59) ve beyazd�r (%90). Hastalar�n %14,7'sinde ba�lang��ta karaci�er metastaz� oldu�u ve �o�u hastan�n (%90) sigara kullanmakta oldu�u veya ge�mi�te kulland��� g�r�lm��t�r. Hastalar�n �o�unun ba�lang�� ECOG performans durumunun 1 oldu�u (%59) ve <%1 oran�nda PD-L1 ekspresyonu sergiledi�i (yakla��k %52) g�r�lm��t�r. �nd�ksiyon tedavisinin ard�ndan yan�t durumu stabil hastal�k, k�smi yan�t veya tam yan�t olan 107 Kol B hastas� aras�nda 40 hasta pemetrexed ge�i� idame tedavisi alm��t�r.
Primer analiz, EGFR mutasyonlar� veya ALK rearanjmanlar� olanlar hari� olmak �zere, ITT- WT olarak tan�mlanan b�t�n hastalarda (n=679) ger�ekle�tirilmi�tir. Hastalarda medyan sa�kal�m takip s�resi 18,6 ay olup, hastalar kontrole k�yasla atezolizumab, nab-paklitaksel ve
karboplatinle OS ve PFS'de iyile�me g�stermi�tir. Temel bulgular, Tablo 6'da �zetlenmi�tir ve OS ve PFS'ye ait Kaplan-Meier e�rileri s�ras�yla �ekil 2 ve 4'te sunulmu�tur. PD-L1 ekspresyonuna g�re OS ve PFS i�in elde edilen ke�ifsel bulgular, s�ras�yla �ekil 3 ve 5'te �zetlenmi�tir. Karaci�er metastazlar� olan hastalar, nab-paklitaksel ve karboplatine k�yasla atezolizumab, nab-paklitaksel ve karboplatinle PFS veya OS'de iyile�me g�stermemi�tir (s�ras�yla PFS i�in 0,93 HR, %95 CI: 0,59, 1,47 ve OS i�in 1,04 HR, %95 CI: 0,63, 1,72).
Atezolizumab, nab-paklitaksel ve karboplatin kolundaki hastalar�n %7,3'�ne kar��l�k nab- paklitaksel ve karboplatin kolundaki hastalar�n %59'u hastal�k progresyonunun ard�ndan �apraz ge�i� tedavisi olarak atezolizumab (b�t�n hastalar�n %41'i) dahil olmak �zere herhangi bir kanser imm�noterapisi alm��t�r.
Daha uzun takip s�resine (medyan: 24,1 ay) sahip bir ke�ifsel analizde, her iki koldaki medyan OS birincil analize g�re de�i�memi� olup, HR=0,82'dir (%95 CI: 0,67, 1,01).
Tablo 6: Primer analizde IMpower130'da kan�tlanan etkilili�in �zeti (ITT-WT pop�lasyon)
Etkililik sonlan�m noktalar� Kol A Atezolizumab + nab-paklitaksel + karboplatin | Kol B Nab- paklitaksel + karboplatin |
Ortak-birincil sonlan�m noktalar� | |
OS n=451 n=228 �l�m say�s� (%) 226 (%50,1) 131 (%57,5) Olaylara kadar ge�en medyan s�re (ay) 18,6 13,9 %95 GA (16, 21,2) (12, 18,7) Grupland�r�lm�� risk oran� (%95 GA) 0,79 (0,64, 0,98) p-de�eri 0,033 12 ayl�k OS (%) 63 56 | |
Ara�t�rmac� taraf�ndan de�erlendirilen PFS (RECIST v1.1) n=451 n=228 Olay say�s� (%) 347 (%76,9) 198 (%86,8) Medyan PFS s�resi (ay) 7 5,5 %95 GA (6,2, 7,3) (4,4, 5,9) Grupland�r�lm�� risk oran� (95% GA) 0,64 (0,54, 0,77) p-de�eri < 0,0001 12 ayl�k PFS (%) %29 %14 | |
Di�er sonlan�m noktalar� |
Ara�t�rmac� taraf�ndan de�erlendirilen ORR (RECIST v1.1)^ n=447 n=226 Do�rulanm�� yan�t veren say�s� (%) 220 (%49,2) 72 (%31,9) %95 GA (44,5, 54) (25,8, 38,4) Tam yan�t say�s� (%) 11 (%2,5) 3 (%1,3) K�smi yan�t say�s� (%) 209 (%46,8) 69 (%30,5) |
Ara�t�rmac� taraf�ndan de�erlendirilen do�rulanm�� DOR (RECIST 1.1)^ n=220 n=72 Medyan (ay olarak) 8,4 6,1 %95 GA (6,9, 11,8) (5,5, 7,9) |
^ Do�rulanm�� ORR ve DoR, ke�ifsel sonlan�m noktalar�d�r
PFS=progresyonsuz sa�kal�m; RECIST=Solid T�m�rlerde Yan�t De�erlendirme Kriterleri v1.1.; GA=g�ven aral���; ORR=objektif yan�t oran�; DOR=yan�t s�resi; OS=Genel sa�kal�m
Ban-pac
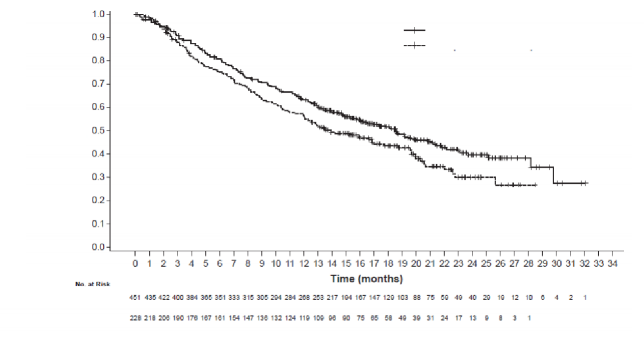
�ekil 2: Genel sa�kal�ma ait Kaplan-Meier e�rileri (IMpower130)
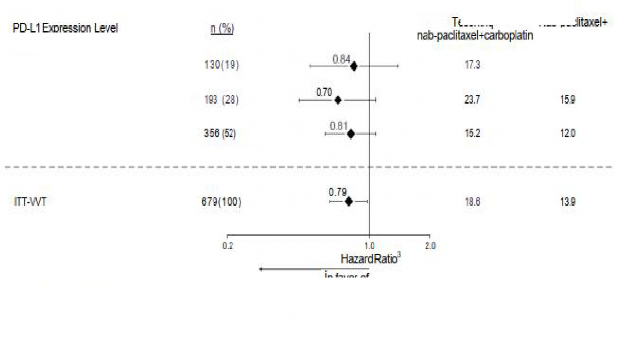
TC > %50 veya IC > %10
TC < %50 ve > %1 veya IC
<%10 ve ≥ %1
TC ve IC < %1
�ekil 3: PD-L1 ekspresyonuna g�re genel sa�kal�m� g�steren meta analiz diyagram� (IMpower130)
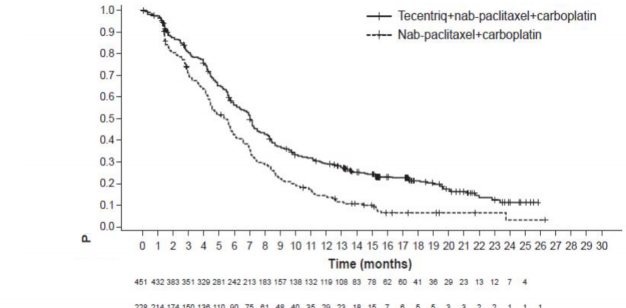
�ekil 4: Progresyonsuz sa�kal�ma ait Kaplan-Meier e�rileri (IMpower130)
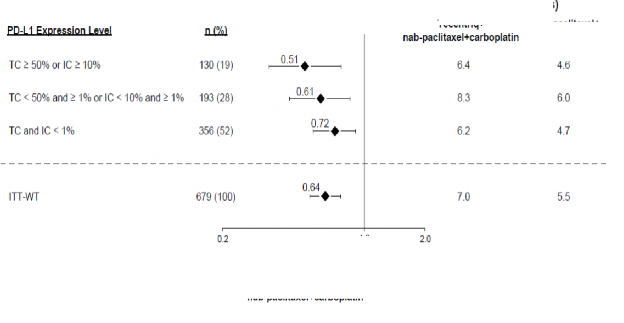
TC≥ %50 veya IC ≥ %10
TC < %50 ve ≥ %1 veya IC
<%10 ve ≥ %1
TC ve IC < %1
�ekil 5: PD-L1 ekspresyonuna g�re progresyonsuz sa�kal�m� g�steren meta analiz diyagram� (IMpower130)
IMpower110 (GO29431): Kemoterapi tedavisi almam�� metastatik KHDAK hastalar�nda yap�lan randomize Faz III �al��ma
Kemoterapi kullanmam��, metastatik KHDAK hastalar�nda atezolizumab�n etkilili�ini ve g�venlili�ini de�erlendirmek i�in Faz III, a��k etiketli, �ok merkezli, randomize bir �al��ma olan IMpower110 y�r�t�lm��t�r. Hastalardaki PD-L1 ekspresyonu ≥%1 TC (PD-L1 t�m�r h�crelerinin ≥%1'i boyanm��t�r) veya ≥%1 IC (t�m�r b�lgesinin ≥%1'ini kapsayan t�m�r- infiltre edici imm�n h�creleri PD-L1 boyanm��t�r) VENTANA PD-L1 (SP142) Testine dayanmaktad�r.
Toplamda 572 hasta randomize edilmi� ve 1:1 oran�nda atezolizumab (A kolu) veya kemoterapi (B kolu) verilmi�tir. Atezolizumab, ara�t�rmac� taraf�ndan de�erlendirildi�i �ekilde klinik fayda kayb�na veya kabul edilmeyen toksisiteye kadar �� haftada bir intraven�z yolla 1200 mg sabit dozla verilmi�tir. Kemoterapi rejimleri Tablo 7'de g�sterilmektedir. Randomizasyon cinsiyet, ECOG performans stat�s�, histoloji ve TC ile IC'de PD-L1 t�m�r ekspresyonu ile katmanla�t�r�lm��t�r.
Tablo 7: Kemoterapi intraven�z tedavi rejimleri (IMpower110)
Tedavi rejimi | �nd�ksiyon (D�rt veya alt� 21 g�nl�k sikluslar) | �dame (21 g�nl�k sikluslar) |
B (Skuamoz olmayan) | Sisplatin (75 mg/m) + pemetreksed (500 mg/m) veya karboplatin (EAA 6) + pemetreksed (500 mg/m) | Pemetreksed (500 mg/m) |
B (Skuamoz) | Sisplatin (75 mg/m) + gemsitabin (1250 mg/m) veya karboplatin (EAA 5) + | En iyi destekleyici bak�m |

gemsitabin (1000 mg/m)
Otoimm�n hastal�k, randomizasyondan �nceki 28 g�n i�inde canl�, zay�flat�lm�� a�� uygulamas�, 4 hafta i�inde sistemik imm�n sistemi uyar�c� ajanlar�n uygulanmas� veya randomizasyondan �nceki 2 hafta i�inde sistemik imm�n sistemi bask�lay�c� ila�lar �yk�s�, aktif veya tedavi edilmemi� merkezi sinir sistemi metastazlar� olan hastalar �al��ma d��� b�rak�lm��t�r. T�m�r de�erlendirmeleri, 1. siklus, 1.g�n ve bunu takip eden ilk 48 hafta boyunca 6 haftada bir ve daha sonra her 9 haftada bir y�r�t�lm��t�r.
EGFR mutasyonlar� veya ALK yeniden d�zenlemeleri olmayan (n=554) PD-L1 ekspresyonu
≥ %1 TC veya ≥ %1 IC olan hastalarda demografik ve �al��ma ba�lang�c� hastal�k �zellikleri tedavi kollar� aras�nda iyi dengelenmi�tir. Medyan ya� 64,5 (da��l�m: 30 ila 87) olup hastalar�n %70'i erkektir. Hastalar�n �o�unlu�u beyaz (%84) ve Asyal�d�r (%14). Hastalar�n �o�u halen sigara kullanmaktad�r veya �ncesinde kullanm��t�r (%87) ve hastalarda ba�lang�� ECOG performans durumu 0 (%36) veya 1'dir (%64).
Genel olarak, hastalar�n %69'unda skuamoz olmayan hastal�k ve hastalar�n %31'inde skuamoz hastal�k vard�r. EGFR mutasyonlar� veya ALK yeniden d�zenlemeleri olmayan (n=205) y�ksek PD-L1 ekspresyonu olan (PD-L1 ≥ %50 TC veya ≥ %10 IC) hastalarda demografik ve �al��ma ba�lang�c� hastal�k �zellikleri genellikle daha geni� �al��ma pop�lasyonunu temsil etmi� ve tedavi kollar� aras�nda dengeli olmu�tur.
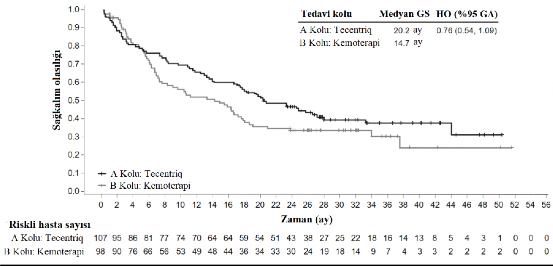
Birincil sonlan�m noktas�, genel sa�kal�m (GS) olmu�tur. Ge�ici genel sa�kal�m analizi s�ras�nda, EGFR mutasyonlar� veya ALK yeniden d�zenlemeleri olanlar (n=205) hari� olmak �zere y�ksek PD-L1 ekspresyonu olan hastalar, kemoterapiye k�yasla atezolizumaba (Kol A) randomize edilen hastalar i�in genel sa�kal�mda istatistiksel olarak anlaml� iyile�me g�stermi� (B kolu) (risk oran� 0,59, %95 GA: 0,4, 0,89; medyan genel sa�kal�m 20,2 aya kar�� 13,1 ay) olup iki tarafl� p de�eri 0,0106 bulunmu�tur. Y�ksek PD-L1 ekspresyonu olan hastalarda ortanca sa�kal�m takip s�resi 15,7 ay olmu�tur.
Bu hastalarda daha uzun bir takip s�resi (medyan: 31,3 ay) i�eren bir ke�ifsel genel sa�kal�m analizinde atezolizumab kolu i�in medyan genel sa�kal�m, birincil genel sa�kal�m ara analizine g�re (20,2 ay) de�i�memi� ve kemoterapi kolu i�in 14,7 olmu�tur (risk oran� 0,76, %95 GA: 0,54, 1,09). Ke�ifsel analizdeki anahtar sonu�lar Tablo 8'de �zetlenmi�tir. Y�ksek PD-L1 ekspresyonu olan hastalarda genel sa�kal�m ve progresyonsuz sa�kal�m i�in Kaplan-Meier e�rileri �ekil 6 ve 7'de sunulmaktad�r. Atezolizumab kolunda (16/107, %15) kemoterapi koluna k�yasla (10/98, %10,2) hastalar�n daha y�ksek bir oran� ilk 2,5 ay i�inde �l�m ya�am��t�r. Erken �l�mlerle ili�kili herhangi bir spesifik fakt�r/fakt�rler tan�mlanamam��t�r.
Tablo 8: Y�ksek PD-L1 ekspresyonu ≥%50 TC veya ≥%10 IC olan hastalarda etkililik �zeti (IMpower110)
Etkililik sonlan�m noktalar� | A Kolu (Atezolizumab) | B Kolu (Kemoterapi) |
Birincil sonlan�m noktas� | ||
Genel sa�kal�m | n=107 | n=98 |
�l�m say�s� (%) | 64 (%59,8) | 64 (%65,3) |
Olaylara kadar ge�en medyan s�re (ay) | 20,2 | 14,7 |
%95 GA | (17,2, 27,9) | (7,4, 17,7) |
S�n�fland�r�lm�� risk oran� (%95 GA) | 0,76 (0,54, 1,09) | |
12 ayl�k genel sa�kal�m (%) | 66,1 | 52,3 |
�kincil sonlan�m noktalar� | ||
Ara�t�rmac� taraf�ndan de�erlendirilen PS (RECIST v1.1) | n=107 | n=98 |
Olay say�s� (%) | 82 (%76,6) | 87 (%88,8) |
Medyan PS s�resi (ay) | 8,2 | 5 |
%95 GA | (6,8, 11,4) | (4,2, 5,7) |
S�n�fland�r�lm�� risk oran� (%95 GA) | 0,59 (0,43, 0,81) | |
12 ayl�k progresyonsuz sa�kal�m (%) | 39,2 | 19,2 |
Ara�t�rmac� taraf�ndan de�erlendirilen OYO (RECIST v1.1)^ | n=107 | n=98 |
Yan�t verenlerin say�s� (%) | 43 (%40,2) | 28 (%28,6) |
%95 GA | (30,8, 50,1) | (19,9, 38,6) |
Tam yan�t say�s� (%) | 1 (%0,9) | 2 (%2) |
K�smi yan�t say�s� (%) | 42 (%39,3) | 26 (%26,5) |
Ara�t�rmac� Taraf�ndan De�erlendirilen YS (RECIST v1.1)^ | n=43 | n=28 |
Medyan (ay) | 38,9 | 8,3 |
%95 GA | (16,1, TE) | (5,6, 11) |
�ekil 6: Y�ksek PD-L1 ekspresyonu ≥%50 TC veya ≥%10 IC olan hastalarda genel sa�kal�m i�in Kaplan-Meier e�risi (IMpower110)
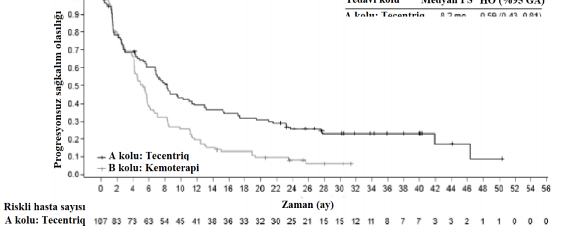
A kolu: Tecentriq 8.2 ay 0.59 (0.43 , 0.81)
B kolu: Kemoterapi 5 ay
�ekil 7: Y�ksek PD-L1 ekspresyonu ≥%50 TC veya ≥%10 IC olan hastalarda progresyonsuz sa�kal�m i�in Kaplan-Meier e�risi (IMpower110)
Kemoterapi koluna k�yasla atezolizumab kolunda g�zlemlenen genel sa�kal�m iyile�mesi, hem skuamoz olmayan k���k h�creli d��� akci�er kanseri hastalar� HR 0,62, %95 GA: 0,4, 0,96; medyan genel sa�kal�m 20,2'ye kar�� 10,5 ay) hem de skuamoz k���k h�creli d��� akci�er kanseri hastalar�n� (HR 0,56, %95 GA: 0,23, 1,37; 15,3 aya kar�� medyan genel sa�kal�ma ula��lamamas�) i�eren alt gruplarda tutarl� bir �ekilde g�sterilmi�tir. 75 ya� ve �zeri hastalara ve hi� sigara i�memi� hastalara ili�kin veriler, bu alt gruplarda sonu� ��karmak i�in �ok s�n�rl�d�r.
�kinci basamak k���k h�creli d��� akci�er kanseri
OAK (GO28915): kemoterapi tedavisi alm�� lokal ileri veya metastatik KHDAK hastalar�nda yap�lan randomize Faz III �al��ma
Platin i�eren bir rejim uygulan�rken veya sonras�nda progresyon g�r�lm��, lokal ileri veya metastatik KHDAK olan hastalarda, atezolizumab ile dosetakselin etkililik ve g�venlilik
kar��la�t�r�lmas�n�n yap�ld��� Faz III, a��k etiketli, �ok merkezli, uluslararas�, randomize bir �al��ma olan GO28915 (OAK) y�r�t�lm��t�r. Bu �al��ma, otoimm�n hastal�k �yk�s� olan, aktif veya kortikosteroid-ba��ml� beyin metastaz� �yk�s� olan, ba�lang��tan �nceki 28 g�n i�inde canl�, atten�e a�� olmu�, ba�lang��tan �nceki 4 hafta i�inde sistemik immunostimulat�r ajan uygulanm�� veya ba�lang��tan �nceki 2 hafta i�inde sistemik immunosupresif t�bbi �r�n kullanm�� hastalar �al��maya al�nmam��t�r. T�m�r de�erlendirmeleri ilk 36 hafta boyunca her 6 haftada bir ve sonra her 9 haftada bir ger�ekle�tirilmi�tir. T�m�r �rnekleri, t�m�r h�crelerinde (TC) ve t�m�r s�zd�ran ba����kl�k h�crelerinde (IC) PD-L1 ifadesi i�in prospektif olarak de�erlendirilmi�tir.
Toplamda 1225 hasta kay�t edilmi�tir ve analiz plan�na g�re randomize edilen ilk 850 hasta primer etki analizine dahil edilmi�tir. Randomizasyon PD-L1 IC ekspresyon durumu, �nceki kemoterapi rejimlerinin say�s� ve histolojiye g�re tabakaland�r�lm��t�r. Hastalar 1:1 oran�nda atezolizumab veya dosetaksel almak �zere randomize edilmi�tir.
Atezolizumab �� haftada bir intraven�z inf�zyon yoluyla 1200 mg sabit dozda uygulanm��t�r. Doz azalt�m�na izin verilmemi�tir. Hastalar, ara�t�rmac� taraf�ndan de�erlendirilecek klinik fayda kayb�na kadar tedavi edilmi�tir. Dosetaksel, progresyona kadar her �� haftal�k siklusun
1. g�n�nde intraven�z inf�zyon yoluyla 75 mg/m uygulanm��t�r. Tedavi edilen t�m hastalar i�in, medyan tedavi s�resi dosetaksel kolu i�in 2,1 ay ve atezolizumab kolu i�in 3,4 ayd�r.
Primer analiz hasta pop�lasyonunun demografik ve ba�lang�� �zellikleri tedavi kollar� aras�nda genel olarak iyi de�erlendirilmi�tir. Medyan ya� 64 ya�t�r (aral�k: 33-85) ve hastalar�n %61'i erkektir. Hastalar�n �o�u beyaz �rktand�r (%70). Hastalar�n yakla��k olarak ��te birinde non- skuamoz hastal�k vard�r (%74). %10'unda EGFR mutasyonu, %0,2'sinde ALK yeniden d�zenlenmesi ve %10'unda ba�lang��ta santral sinir sistemi metastazlar� vard�r. Hastalar�n �o�u halen veya �nceden t�t�n kullan�c�s�d�r (%82). Ba�lang��ta ECOG performans skoru 0 (%37) veya 1'dir (%63). Hastalar�n %75'i �nceden yaln�zca bir platin bazl� tedavi rejimi alm��t�r.
Birincil etkililik sonlan�m noktas�, randomizasyon tarihinden herhangi bir nedenle �l�me kadar ge�en s�re olarak tan�mlanan GS'dir (genel sa�kal�m). 21 ayl�k medyan sa�kal�m takip s�resini i�eren bu �al��man�n anahtar sonu�lar� Tablo 9'da �zetlenmi�tir. ITT pop�lasyonu (tedavi ama�l�) i�in Kaplan-Meier e�rileri �ekil 8'de sunulmu�tur. �ekil 9, ITT ve PD-L1 alt gruplar�ndaki GS sonu�lar�n� �zetlemekte olup , TC ve IC'de % 1'in alt�nda PD-L1 ekspresyonu olanlar� da i�eren t�m alt gruplarda atezolizumab ile GS faydas�n� g�stermektedir.
Tablo 9: OAK �al��mas�ndan primer analiz pop�lasyonunda etkililik �al��mas� (t�m gelenler)*
Etkililik sonlan�m noktas� | Atezolizumab (n=425) | Dosetaksel (n=425) |
Birincil Etkililik Sonlan�m Noktas� |
|
|
GS |
|
|
�l�m say�s� (%) | 271 (%64) | 298 (%70) |
Olaylara kadar ge�en medyan s�re (ay) | 13,8 | 9,6 |
%95 GA | (11,8; 15,7) | (8,6; 11,2) |
S�n�fland�r�m�� risk oran� (%95 GA) | 0,73 (0,62; 0,87) | |
p de�eri** | 0,0003 |
|
Etkililik sonlan�m noktas� | Atezolizumab (n=425) | Dosetaksel (n=425) |
12 ayl�k GS*** | 218 (%55) | 151 (%41) |
18 ayl�k GS*** | 157 (%40) | 98 (%27) |
�kincil Etkililik Sonlan�m Noktalar� |
|
|
Ara�t�rmac� taraf�ndan de�erlendirilen PS (RECIST v1.1) | ||
Olay say�s� (%) | 380 (%89) | 375 (%88) |
Medyan PS s�resi (ay) | 2,8 | 4 |
%95 GA | (2,6; 3) | (3,3; 4,2) |
S�n�fland�r�lm�� risk oran� (%95 GA) | 0,95 (0,82; 1,1) | |
Ara�t�rmac� Taraf�ndan De�erlendirilen OYO (RECIST v1.1) | ||
Yan�t verenlerin say�s� (%) | 58 (%14) | 57 (%13) |
%95 GA | (10,5; 17,3) | (10,3;17) |
Ara�t�rmac� Taraf�ndan De�erlendirilen YS (RECIST v1.1) | ||
| n=58 | n=57 |
Medyan (ay) | 16,3 | 6,2 |
%95 GA | (10; TE) | (4,9; 7,6) |
GA=g�ven aral���; YS=yan�t s�resi; TE=Tahmin edilemiyor; OYO=objektif yan�t oran�; GS=genel sa�kal�m; PS=progresyonsuz sa�kal�m; RECIST-STCDK=Solid T�m�rlerde Cevap De�erlendirme Kriterleri v1.1.
*Primer analiz pop�lasyonu randomize edilen ilk 850 hastay� i�erir
†IC d�zeyleri, t�m�r s�zd�ran ba����kl�k h�crelerindeki PD-L1 ekspresyonuna, �nceki kemoterapi rejimi say�s� ve histolojiye g�re s�n�fland�r�lm��t�r.
** s�n�fland�r�lm�� log-s�ra s�ralamas�na g�re
*** Kaplan-Meier hesaplamalar�na g�re
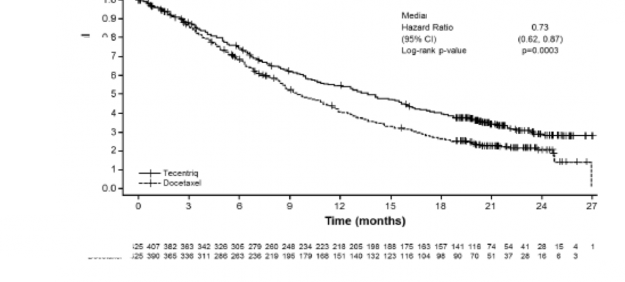
Zaman (ay)
Sa�kal�m olas�l���
�ekil 8: Primer analiz pop�lasyonunda genel sa�kal�m i�in Kaplan-Meier e�risi (t�m gelenler) (OAK)
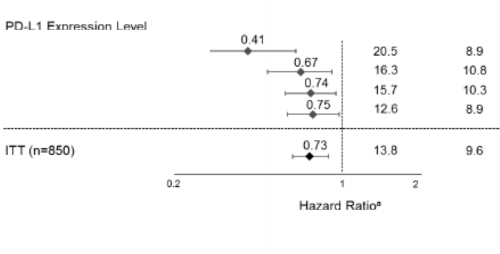
�ekil 9: Primer analiz pop�lasyonunda PD-L1 ekspresyonuna g�re genel sa�kal�m meta- analiz diyagram� (OAK)
PD-L1 Ekspresyon Seviyesi
TC≥ %50 veya IC ≥ %10 (n=137) TC veya IC ≥ %5 (n=265)
TC veya IC ≥ %1 (n=463) TC ve IC < %1 (n=379)
Medyan GS (ay) TECENTRIQ Dosetaksel
TECENTRIQ
lehine
TC ve IC ≥ % 1i�in s�n�fland�r�lm�� HR. Di�er a��klay�c� altgruplar i�i s�n�fland�r�lmam�� HR
TC: T�m�r h�creleri; IC: �mm�n h�creleri; HR: Risk oran�
Hem non-skuamoz KHDAK hastalar�nda (atezolizumab and dosetaksel i�in s�ras�yla; risk oran� [HR]: 0,73, 95% GA: 0,6, 0,89; medyan GS< 15,6'ya kar�� 11,2 ay) hem de skuamoz KHDAK
hastalar�nda (atezolizumab and dosetaksel i�in s�ras�yla HR: 0,73, 95% GA: 0,54, 0,98; medyan GS: 8,9 vs. 7,7 ay) dosetaksel ile kar��la�t�r�lan GS de�erlerinde geli�me g�r�lm��t�r. G�zlemlenen GS geli�imi, ba�lang��ta beyin metastaz� olanlar (atezolizumab and dosetaksel i�in s�ras�yla; risk oran� [HR]: 0,54, 95% GA: 0,31, 0,94; medyan GS< 20,1 vs. 11,9 ay) ve hi� t�t�n t�ketmemi� olanlar (atezolizumab and dosetaksel i�in s�ras�yla; risk oran� [HR]: 0,71, 95% GA: 0,47, 1,08; medyan GS< 16,3 vs. 12,6 ay) dahil olmak �zere altgruplar aras�nda tutarl� bir bi�imde g�sterilmi�tir. Bununla birlikte, EGFR mutasyonlu hastalar dosetaksele kar��l�k atezolizumab kullan�m�nda geli�mi� GS g�stermemi�lerdir (atezolizumab ve dosetaksel i�in s�ras�yla; risk oran� [HR]: 1,24, 95% GA: 0,71, 2,18; medyan GS< 10,5 ‘e kar�� 16,2 ay).
EORTC QLQ-LC13 ile �l��ld���nde, hasta bildirimli g���s a�r�s�ndaki k�t�le�meye kadar zaman�n, dosetaksele kar��l�k atezolizumab i�in (HR: 0,71, 95% GA: 0,49, 1,05; iki kolda da medyana ula��lmam��t�r) uzad��� g�r�lm��t�r. EORTC QLQ-LC13 ile �l��len di�er akci�er kanseri semptomlar� (�rn. �ks�r�k, dispne ve kol/omuz a�r�s�) i�in k�t�le�me s�releri atezolizumab ve dosetaksel i�in benzerdir. Bu sonu�lar �al��man�n a��k etiketli dizayn�na dayanarak dikkatle yorumlanm��t�r.
POPLAR (GO28753): kemoterapi tedavisi alm�� lokal ileri veya metastatik KHDAK hastalar�nda yap�lan randomize Faz II �al��ma
PD-L1 ekspresyonundan ba��ms�z olarak platin i�eren rejim uygulan�rken ya da sonras�nda progresyon g�r�lm��, lokal ileri veya metastatik KHDAK olan hastalarda Faz II, �ok merkezli, uluslararas�, randomize, a��k etiketli, kontroll� bir �al��ma olan GO28753 (POPLAR) �al��mas� da y�r�t�lm��t�r. Birincil etkililik sonlan�m noktas�, genel sa�kal�md�r. Toplam 287 hasta 1:1 oran�nda TECENTRIQ (klinik fayda kayb�na kadar �� haftada bir intraven�z inf�zyon yoluyla 1200 mg) ya da dosetaksel (progresyona kadar her �� haftal�k siklusun 1. (birinci) g�n�nde intraven�z inf�zyon yoluyla 75 mg/m) almak �zere randomize edilmi�tir. Randomizasyon, PD-L1 IC ekspresyon durumu, �nceki kemoterapi rejimlerinin say�s� ve histolojiye g�re
tabakaland�r�lm��t�r.
G�zlemlenen toplam 200 �l�m ve 22 ayl�k medyan sa�kal�m takibi ile g�ncelle�tirilmi� analiz; dosetaksel ile tedavi edilen hastalarda medyan sa�kal�m 9,7 ay ve atezolizumab ile tedavi edilen hastalarda medyan sa�kal�m 12,6 ay (HR: 0,69% 95 GA: 0,52, 0,92) olarak g�stermi�tir. Atezolizumab ve dosetaksel i�in Objektif Yan�t Oran� (OYO) s�ras�yla %15,3'e kar�� %14,7, medyan Yan�t S�resi (YS) ise 18,6 ay ve 7,2 ayd�r.
K���k h�creli akci�er kanseri:
IMpower133 (GO30081): kemoterapi tedavisi almam�� yayg�n evre KHAK hastalar�nda karboplatin ve etoposidle kombinasyon halinde yap�lan randomize Faz I/III �al��ma
Kemoterapi kullanmam��, yayg�n evre KHAK hastalar�nda atezolizumab�n karboplatin ve etoposid ile kombinasyon halinde kullan�ld���nda etkilili�ini ve g�venlili�ini de�erlendirmek i�in Faz I/III, randomize, �ok merkezli, �ift-k�r, plasebo kontroll� bir �al��ma olan IMpower133 y�r�t�lm��t�r.
Aktif veya tedavi edilmemi� beyin metastaz� olan, otoimm�n hastal�k �yk�s� olan, randomizasyondan �nceki 4 hafta i�inde canl�, atten�e a�� olmu�, randomizasyondan �nceki 1 (bir) hafta i�inde sistemik immunostimulat�r ajan uygulanm�� hastalar �al��maya al�nmam��t�r.
T�m�r de�erlendirmeleri 1. (birinci) siklusun 1. (birinci) g�n�n� takiben ilk 48 hafta boyunca her 6 haftada bir ve sonra her 9 haftada bir ger�ekle�tirilmi�tir. Hastal�k progresyonundan sonra tedavi edilen hastalar�n t�m�r de�erlendirmeleri kullan�m�n b�rak�lmas�na kadar her 6 haftada bir ger�ekle�tirilmi�tir.
Toplamda 403 hasta kay�t edilmi�tir ve 1:1 oran�nda Tablo 10'daki tedavi rejimlerinden birini almak �zere randomize edilmi�tir. Randomizasyon cinsiyet, ECOG performans skoru ve beyin metastaz varl���na g�re s�n�fland�r�lm��t�r.
Tablo 10: Intraven�z tedavi rejimi (IMpower133)
Tedavi rejimi | �nd�ksiyon (21-g�nl�k 4 siklus) | �dame (21-g�nl�k siklus) |
A | atezolizumab (1200 mg) + karboplatin (EAA 5) ve etoposid (100 mg/m) | atezolizumab (1200 mg) |
B | plasebo + karboplatin (EAA 5) ve etoposid (100 mg/m) | plasebo |
Hasta pop�lasyonunun demografik ve ba�lang�� �zellikleri tedavi kollar� aras�nda genel olarak benzer de�erlendirilmi�tir. Hastalar�n %10'u 75 ya� ve �zeri olmakla beraber medyan ya� 64 y�ld�r (aral�k: 26-90 ya�). Hastalar�n �o�unlu�u erkektir (%65), beyaz �rktand�r (%80), %9'unda beyin metastaz� vard�r ve �o�u halen veya �nceden t�t�n kullan�c�s�d�r (%97). Ba�lang��ta
ECOG performans skoru 0 (%35) veya 1(bir)'dir (%65).
Primer analizde, hastalar�n medyan sa�kal�m takip s�resi 13,9 ayd�r. Kontrol koluna k�yasla atezolizumab ile karboplatin ve etoposid kombinasyonu ile genel sa�kal�mda istatistiksel olarak anlaml� bir geli�me g�zlendi (risk oran� (HR) 0,7 %95 GA: 0,54, 0,91; ortalama genel sa�kal�m : 12,3 aya 10,3 ay). Uzun d�nem takipli ara�t�rma genel sa�kal�m analizinde (ortalama: 22,9 ay), her iki koldaki ortalama genel sa�kal�m, ba�lang�� genel sa�kal�m ara analizine g�re de�i�memi�tir. Ara�t�rma analizi yan�nda ba�lang�� analiziden elde edilen Progresyonsuz Sa�kal�m (PS), Objektif Yan�t Oran� (OYO)ve Yan�t S�resi (YS) sonu�lar� Tablo 11'de �zetlenmi�tir. Genel sa�kal�m ve progresyonsuz sa�kal�m sonu�lar�n� g�steren Kaplan-Meier e�rileri �ekil 10'da ve 11'de sunulmu�tur. Beyin metastaz� olan hastalar i�in veriler bu pop�lasyonla ilgili sonu� ��karmak i�in �ok s�n�rl�d�r.
Tablo 11: Etkililik �zeti (IMpower133)
Etkililik sonlan�m noktas� | A kolu (atezolizumab + karboplatin + etoposid) | B kolu (plasebo + karboplatin + etoposid) |
E� Sonlan�m Noktalar� | ||
GS analizi | n=201 | n=202 |
�l�m say�s� (%) | 142 (%70,6) | 160 (%79,2) |
Olaylara kadar ge�en medyan s�re (ay) | 12,3 | 10,3 |
%95 GA | (10,8; 15,8) | (9,3; 11,3) |
Tabakaland�r�lm�� risk oran� (%95 GA) | 0,76 (0,6; 0,95) | |
p de�eri |
0,0154*** | |
12 ayl�k GS (%) | 51,9 | 39 |
Ara�t�rmac� taraf�ndan de�erlendirilen PS (RECIST-STCDK v1.1)** ^ | n=201 | n=202 |
Olay say�s� (%) | 171 (%85,1) | 189 (%93,6) |
Medyan PS s�resi (ay) | 5,2 | 4,3 |
%95 GA | (4,4; 5,6) | (4,2; 4,5) |
S�n�fland�r�lm�� risk oran� (%95 GA) | 0,77 (0,62; 0,96) | |
p de�eri | 0,017 |
|
6 ayl�k PS (%) | 30,9 | 22,4 |
12 ayl�k PS (%) | 12,6 | 5,4 |
Di�er sonlan�m noktalar� | ||
Ara�t�rmac� Taraf�ndan De�erlendirilen OYO (RECIST-STCDK v1.1)** ^ | n=201 | n=202 |
Yan�t verenlerin say�s� (%) | 121 (%60,2) | 130 (%64,4) |
%95 GA | (53,1; 67) | (57,3; 71) |
Tam yan�t say�s� (%) | 5 (%2,5) | 2 (%1) |
Parsiyel yan�t say�s� (%) | 116 (%57,7) | 128 (%63,4) |
Ara�t�rmac� Taraf�ndan De�erlendirilen YS (RECIST-STCDK v1.1)** ^ | n=121 | n=130 |
Etkililik sonlan�m noktas� | A kolu (atezolizumab + karboplatin + etoposid) | B kolu (plasebo + karboplatin + etoposid) |
Medyan (ay) | 4,2 | 3,9 |
%95 GA | (4,1; 4,5) | (3,1; 4,2) |
PS=progresyonsuz sa�kal�m; RECIST-STCDK=Solid T�m�rlerde Cevap De�erlendirme Kriterleri v1.1.; GA=g�ven aral���; OYO=objektif yan�t oran�; YS=objektif yan�t s�resi;
GS=genel sa�kal�m
* Ara�t�rma GS final analizi klinik kesim tarihi 21 Ocak 2019
**PS, OYO ve YS analizi klinik kesim tarihi 24 Nisan 2018
*** Sadece tan�mlay�c� ama�lar i�in
^ Onaylanan OYO ve YS ara�t�rma sonlan�m noktas�d�r
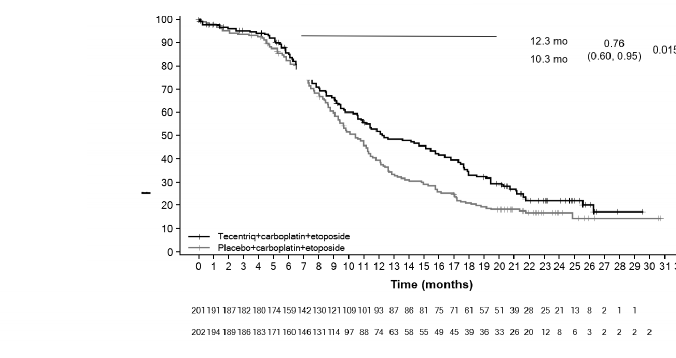
Tedavi kolu
Medyan GS HR (%95 GA) p-de�eri
Genel sa�kal�m (%)
�ekil 10: Genel sa�kal�m i�in Kaplan-Meier e�risi (IMpower133)
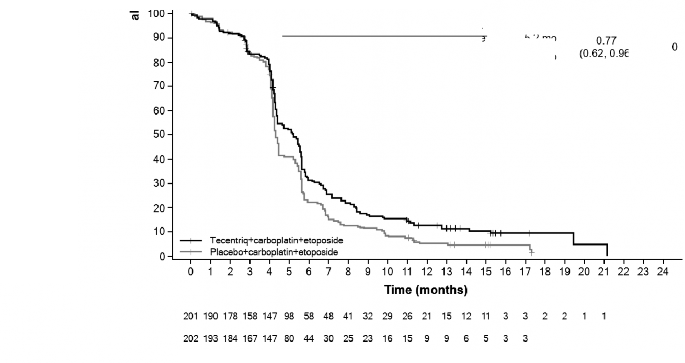
Progresyonsuz sa�kal�m (%)
�ekil 11: Progresyonsuz sa�kal�m i�in Kaplan-Meier e�risi (IMpower133)
�rotelyal Kanser:
IMvigor211 (GO29294): Daha �nce kemoterapi ile tedavi edilmi� lokal ileri veya metastatik evre �rotelyal kanserli hastalar �zerinde yap�lan randomize �al��ma
Platin i�eren bir rejim s�ras�nda veya sonras�nda hastal�k ilerlemesi g�r�len lokal ileri veya metastatik evre �rotelyal kanserli hastalarda kemoterapiye (ara�t�rmac�n�n se�imine ba�l� olarak vinflunin, dosetaksel veya paklitaksel) k�yasla atezolizumab�n etkilili�ini ve g�venlili�ini de�erlendirmek i�in faz III, a��k etiketli, �ok merkezli, uluslararas�, randomize bir �al��ma (IMvigor211) y�r�t�lm��t�r. Otoimm�n hastal�k �yk�s� olan, aktif veya kortikosteroid-ba��ml� beyin metastaz� olan, kay�ttan �nceki 28 g�n i�inde canl�, atten�e a�� uygulanm�� ve kay�ttan �nceki 4 (d�rt) hafta i�inde sistemik immunostimulat�r ajan veya 2 (iki) hafta i�inde sistemik immunosupresif t�bbi �r�n uygulanm�� hastalar bu �al��madan d��lanm��t�r. T�m�r de�erlendirmeleri ilk 54 hafta boyunca her 9 (dokuz) haftada bir ve sonras�nda her 12 haftada bir ger�ekle�tirilmi�tir. T�m�r �rnekleri, t�m�r� infiltre eden imm�n h�crelerde (IC) PD-L1 ekspresyonu i�in prospektif olarak de�erlendirilmi� ve sonu�lar a�a��da a��klanan analizler i�in PD-L1 ekspresyon alt gruplar�n� tan�mlamak i�in kullan�lm��t�r.
Toplam 931 hasta kaydedilmi�tir. Hastalar atezolizumab ya da kemoterapi almak �zere randomize edilmi�tir (1 (bir):1(bir)). Randomizasyon kemoterapi (vinflunin ya da taksan), IC'de PD-L1 ekspresyon durumu (<%5 (be�)'e kar�� ≥%5 (be�)), prognostik risk fakt�rlerinin say�s� (0'a kar�� 1(bir)-3 (��)) ve karaci�er metastaz�na (evete kar�� hay�r) g�re s�n�fland�r�lm��t�r. Prognostik risk fakt�rleri aras�nda �nceki kemoterapiden itibaren 3 (��) ay ge�mesi, 0 ECOG performans durumu ve 10 g/dL hemoglobin yer alm��t�r.
Atezolizumab, 3 (��) haftada bir intraven�z inf�zyon yoluyla 1200 mg sabit dozda
uygulanm��t�r. Atezolizumab dozunun azalt�m�na izin verilmemi�tir. Hastalar, ara�t�rmac� taraf�ndan de�erlendirildi�i �ekilde klinik faydan�n kaybedilmesine veya kabul edilemez toksisite g�r�lene kadar tedavi edilmi�tir. Vinflunin, hastal���n ilerlemesine veya kabul edilemez toksisite g�r�lene kadar her 3 (��) haftal�k d�ng�n�n 1. (birinci) g�n�nde intraven�z inf�zyon yoluyla 320 mg/m dozunda uygulanm��t�r. Paklitaksel, hastal���n ilerlemesine veya kabul edilemez toksisite g�r�lene kadar her 3 (��) haftal�k d�ng�n�n 1. (birinci) g�n�nde 3 (��) saat boyunca intraven�z inf�zyon yoluyla 175 mg/m dozunda uygulanm��t�r. Dosetaksel, hastal���n ilerlemesine veya kabul edilemez toksisite g�r�lene kadar her 3 (��) haftal�k d�ng�n�n 1. (birinci) g�n�nde intraven�z inf�zyon yoluyla 75 mg/m dozunda uygulanm��t�r. Tedavi edilen t�m hastalarda medyan tedavi s�resi atezolizumab kolu i�in 2,8 ay, vinflunin ve paklitaksel kollar� i�in 2,1 ay ve dosetaksel kolu i�in 1,6 ay olmu�tur.
Primer analiz pop�lasyonunun demografik ve ba�lang�� hastal�k �zellikleri tedavi kollar� aras�nda dengeli olmu�tur. Medyan ya� 67 (aral�k: 31-88) olup hastalar�n %77,1'i erkektir. Hastalar�n �o�unlu�u (%72,1) beyaz �rktan olup kemoterapi kolundaki hastalar�n %53,9'u vinflunin alm��, ba�lang��ta hastalar�n %71,4'�nde en az bir k�t� prognostik risk fakt�r� ve %28,8'inde karaci�er metastaz� mevcuttur. Ba�lang�� ECOG performans durumu 0 (%45,6) veya 1 (%54,4) olmu�tur. Hastalar�n %71,1'inde primer t�m�r yeri mesanedir ve hastalar�n %25,4'�nde �st sistem �rotelyal karsinomu mevcuttur. Hastalar�n %24,2'si daha �nce sadece platin i�eren adjuvan veya neoadjuvan tedavi alm�� ve bu hastalarda 12 ay i�inde hastal�k ilerlemesi g�r�lm��t�r.
IMvigor211 i�in primer etkililik sonlan�m noktas� genel sa�kal�md�r (GS). Ara�t�rmac� taraf�ndan de�erlendirilen Solid T�m�rlerde Yan�t De�erlendirme Kriterleri (RECIST) v1.1'e g�re de�erlendirilen sekonder etkililik sonlan�m noktalar� objektif yan�t oran� (OYO), progresyonsuz sa�kal�m (PS) ve yan�t s�residir (YS). IC2/3, IC1/2/3ve ITT (tedavisi ama�lanan, yani t�m kat�lanlar) pop�lasyonlar�nda tedavi kolu ve kontrol kolu aras�ndaki GS kar��la�t�rmalar�, a�a��daki �ekilde %5'lik iki tarafl� seviyede s�n�fland�r�lm�� bir log-s�ra testine dayal� hiyerar�ik, sabit s�ral� bir prosed�r kullan�larak test edilmi�tir: a�ama 1) IC2/3pop�lasyonu; a�ama 2) IC1/2/3 pop�lasyonu; a�ama 3) t�m kat�lanlar pop�lasyonu. A�ama 2 ve 3'�n her birine ait GS sonu�lar�, ancak �nceki a�amaya ait sonu� istatistiksel olarak anlaml�ysa, istatistiksel anlaml�l�k a��s�ndan resmi olarak test edilebilmi�tir.
Medyan sa�kal�m takibi 17 ayd�r. IMvigor211 �al��mas�n�n primer analizi primer genel sa�kal�m sonlan�m noktas�n� kar��lamam��t�r. Atezolizumab, daha �nce tedavi edilmi�, lokal ileri evre veya metastatik �rotelyal karsinomlu hastalarda kemoterapiye k�yasla istatistiksel olarak anlaml� bir sa�kal�m faydas� g�stermemi�tir. �nceden belirlenmi� hiyerar�ik test s�ras�na g�re, 0,87'lik bir genel sa�kal�m risk oran� ile �nce IC 2/3pop�lasyonu test edilmi�tir (%95 GA: 0,63, 1,21; atezolizumab ve kemoterapi i�in medyan genel sa�kal�m s�ras�yla 11,1 aya kar�� 10,6 ay). S�n�fland�r�lm�� log-s�ra p de�eri 0,41'dir ve bu nedenle sonu�lar bu pop�lasyonda istatistiksel olarak anlaml� kabul edilmemi�tir. Sonu� olarak, IC1/2/3 veya t�m kat�lanlar pop�lasyonunda genel sa�kal�m i�in herhangi bir istatistiksel anlaml�l�k testi ger�ekle�tirilememi� ve bu analizlerin sonu�lar� a��klay�c� olarak de�erlendirilmi�tir. T�m kat�lanlar pop�lasyonundaki kilit sonu�lar Tablo 12'de �zetlenmi�tir. T�m kat�lanlar pop�lasyonunda Kaplan Meier genel sa�kal�m e�risi �ekil 12'de sunulmaktad�r.
ITT pop�lasyonunda 34 ayl�k bir medyan sa�kal�m takip s�resi ile a��klay�c� bir g�ncellenmi� sa�kal�m analizi ger�ekle�tirilmi�tir. Medyan genel sa�kal�m, 0,82'lik bir risk oran� (%95 GA: 0,71, 0,94) ile atezolizumab kolunda 8,6 ay (%95 GA: 7,8, 9,6) ve kemoterapi kolunda 8 ay (%95 GA: 7,2, 8,6) olmu�tur. 12 ayl�k genel sa�kal�m oranlar� i�in primer analizde g�zlemlenen e�ilim ile uyumlu olarak, atezolizumab kolundaki hastalarda, ITT pop�lasyonundaki
kemoterapi koluna k�yasla say�sal olarak daha y�ksek 24 ve 30 ayl�k genel sa�kal�m oranlar� g�zlemlenmi�tir. 24. ayda sa� olan hastalar�n y�zdesi (KM tahmini) kemoterapi kolunda %12,7 ve atezolizumab kolunda %22,5; 30. ayda sa� olan hastalar�n y�zdesi (KM tahmini) ise kemoterapi kolunda %9,8 ve atezolizumab kolunda %18,1 olmu�tur.
Tablo 12: T�m kat�lanlarda etkililik �zeti (IMvigor211)
Etkililik sonlan�m noktas� | Atezolizumab (n = 467) | Kemoterapi (n = 464) |
Primer etkililik sonlan�m noktas� | ||
GS* | ||
�l�m say�s� (%) | 324 (%69,4) | 350 (%75,4) |
Olaylara kadar ge�en medyan s�re (ay) | 8,6 | 8 |
%95 GA | 7,8, 9,6 | 7,2, 8,6 |
S�n�fland�r�lm�� risk oran� (%95 GA) | 0,85 (0,73, 0,99) | |
12 ayl�k GS (%)** | %39,2 | %32,4 |
Sekonder ve a��klay�c� sonlan�m noktalar� | ||
Ara�t�rmac� taraf�ndan de�erlendirilen PS (RECIST v1.1) | ||
Olay say�s� (%) | 407 (%87,2) | 410 (%88,4) |
Medyan PS s�resi (ay) | 2,1 | 4 |
%95 GA | 2,1, 2,2 | 3,4, 4,2 |
S�n�fland�r�lm�� risk oran� (%95 GA) | 1,1 (0,95, 1,26) | |
Ara�t�rmac� taraf�ndan de�erlendirilen OYO (RECIST v1.1) | n = 462 | n = 461 |
Do�rulanm�� yan�t verenlerin say�s� (%) | 62 (%13,4) | 62 (%13,4) |
%95 GA | 10,45, 16,87 | 10,47, 16,91 |
Tam yan�t say�s� (%) | 16 (%3,5) | 16 (%3,5) |
K�smi yan�t say�s� (%) | 46 (%10) | 46 (%10) |
Stabil hastal�k say�s� (%) | 92 (%19,9) | 162 (%35,1) |
Ara�t�rmac� taraf�ndan de�erlendirilen YS (RECIST v1.1) | n = 62 | n = 62 |
Medyan (ay) *** | 21,7 | 7,4 |
%95 GA | 13, 21,7 | 6,1, 10,3 |
GA=g�ven aral���; YS=yan�t s�resi; OYO=objektif yan�t oran�; GS=genel sa�kal�m; PS=progresyonsuz sa�kal�m; RECIST = Kat� T�m�rlerde Yan�t De�erlendirme Kriterleri v1.1.
* T�m kat�lanlar pop�lasyonunda GS analizi s�n�fland�r�lm�� log-s�ra testine g�re ger�ekle�tirilmi� ve sonu� sadece a��klama ama�l� olarak sunulmu�tur (p = 0,0378); �nceden belirlenmi� analiz hiyerar�isine g�re, t�m kat�lanlar pop�lasyonunda GS analizi i�in p de�eri istatistiksel olarak anlaml� kabul edilememektedir.
ǂ Kemoterapi (vinflunin ya da taksan), IC'de durum (< %5 (be�)'e kar�� ≥ %5(be�)), prognostik risk fakt�rlerinin say�s� (0'a kar�� 1(bir)-3(��)) ve karaci�er metastaz�na (evete kar�� hay�r) g�re s�n�fland�r�ld�.
** Kaplan-Meier tahminine g�re.
*** Yan�tlar atezolizumab kolunda yan�t verenlerin %63'�nde ve kemoterapi kolunda yan�t verenlerin %21'inde devaml�yd�.
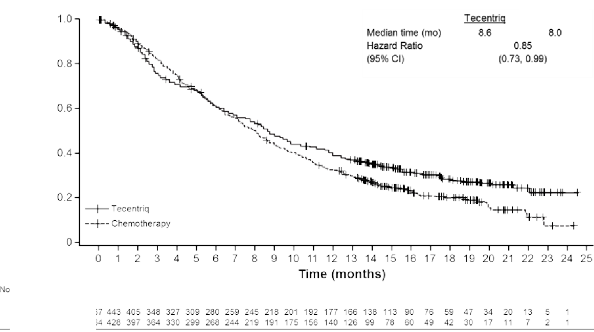
�ekil 12: Kaplan Meier genel sa�kal�m e�risi (IMvigor211)
IMvigor210 (GO29293): Sisplatin tedavisi i�in uygun bulunmayan, daha �nceden tedavi edilmemi� �rotelyal kanserli hastalar �zerinde yap�lan tek kollu �al��ma
Lokal ileri veya metastatik evre �rotelyal kanserli (�rotelyal mesane kanseri olarak da bilinir) hastalar �zerinde faz II, �ok merkezli, uluslararas�, iki kohortlu, tek kollu bir klinik �al��ma (IMvigor210) y�r�t�lm��t�r.
�al��mada toplam 438 hasta ve iki hasta kohortu vard�r. Kohort 1'e, sisplatin bazl� kemoterapiye uygun bulunmayan veya uygun olmayan ya da platin i�eren bir neoadjuvan veya adjuvan kemoterapi rejimi ile uygulanan tedaviden en az 12 ay sonra hastal�k ilerlemesi g�r�len, daha �nce tedavi g�rmemi�, lokal ileri veya metastatik evre �rotelyal kanserli hastalar dahil edilmi�tir. Kohort 2'ye ise, lokal ileri veya metastatik evre �rotelyal kanserli i�in en az bir platin bazl� kemoterapi rejimi alan veya platin i�eren bir neoadjuvan veya adjuvan kemoterapi rejimi ile uygulanan tedaviden sonraki 12 ay i�inde hastal�k ilerlemesi g�r�len hastalar dahil edilmi�tir.
Kohort 1'de, 119 hasta hastal���n ilerlemesine kadar intraven�z inf�zyon yoluyla 3 (��) haftada bir 1200 mg atezolizumab ile tedavi edilmi�tir. Medyan ya� 73't�r. Hastalar�n �o�u (%81) erkek ve �o�unlu�u (%91) beyaz �rktan olmu�tur.
Kohort 1'e, ECOG performans durumu 0 olan 45 hasta (%38), ECOG performans durumu 1 olan 50 hasta (%42) ve ECOG performans durumu 2 olan 24 hasta (%20), Bajorin risk fakt�r� (ECOG performans durumu ≥ 2 ve viseral metastaz) olmayan 35 hasta (%29), bir Bajorin risk fakt�r� olan 66 hasta (%56) ve iki Bajorin risk fakt�r� olan 18 hasta (%15), b�brek i�lev bozuklu�u olan (glomer�ler filtrasyon h�z� [GFR] < 60 mL/dk) 84 hasta (%71) ve karaci�er metastaz� olan 25 hasta (%21) dahil edilmi�tir.
Kohort 1'in primer etkililik sonlan�m noktas�, ba��ms�z bir de�erlendirme tesisi (IRF) taraf�ndan RECIST v1.1 kullan�larak de�erlendirilen objektif yan�t oran� (OYO) olarak do�rulanm��t�r.
Primer analiz, t�m hastalar en az 24 hafta s�reyle takip edildi�inde ger�ekle�tirilmi�tir. T�m kat�lanlarda medyan tedavi s�resi 15 hafta ve medyan sa�kal�m takip s�resi 8,5 ay olmu�tur. RECIST v1.1'e g�re IRF taraf�ndan de�erlendirilen klinik olarak anlaml� OYO'lar g�sterilmi�; ancak �nceden belirlenmi� %10'luk bir ge�mi� kontrol yan�t oran� ile kar��la�t�r�ld���nda, anlaml� faydaya ula��lamam��t�r. IRF RECIST v1.1'e g�re do�rulanm�� OYO'lar PD-L1 ekspresyonu ≥%5 olan hastalarda %21,9 (%95 GA: 9,3, 40), PD-L1 ekspresyonu ≥%1 olan
hastalarda %18,8 (%95 GA: 10,9, 29) ve t�m kat�lanlarda %19,3 (%95 GA: 12,7, 27,6) olmu�tur. Herhangi bir PD-L1 ekspresyon alt grubunda veya t�m kat�lanlarda medyan yan�t s�resine (YS) ula��lmam��t�r. Yakla��k %40'l�k bir olay-hasta oran� ile genel sa�kal�m mat�r de�ildir. T�m hasta alt gruplar�nda (PD-L1 ekspresyonu %5 ve %1) ve t�m kat�lanlarda medyan genel sa�kal�m 10,6 ay olmu�tur.
Kohort 1 i�in ger�ekle�tirilen ve 17,2 ayl�k bir medyan sa�kal�m takip s�resi ile g�ncellenmi� olan analiz Tablo 13'te �zetlenmektedir. Herhangi bir PD-L1 ekspresyon alt grubunda veya t�m kat�lanlarda medyan YS'ye ula��lmam��t�r.
Tablo 13: G�ncellenmi� etkililik �zeti (IMvigor210 Kohort 1)
Etkililik sonlan�m noktas� | IC'de PD-L1 ekspresyonu ≥%5 olanlar | IC'de PD-L1 ekspresyonu ≥%1 olanlar |
T�m Kat�lanlar |
OYO (IRF taraf�ndan de�erlendirilen; RECIST v1.1) | n = 32 | n = 80 | n = 119 |
Yan�t verenlerin say�s� (%) | 9 (%28,1) | 19 (%23,8) | 27 (%22,7) |
%95 GA | 13,8, 46,8 | 15, 34,6 | 15,5, 31,3 |
Tam yan�t say�s� (%) %95 GA | 4 (%12,5) (3,5, 29) | 8 (%10) (4,4, 18,8) | 11 (%9,2) (4,7, 15,9) |
K�smi yan�t say�s� (%) %95 GA | 5 (%15,6) (5,3, 32,8) | 11 (%13,8) (7,1, 23,3) | 16 (%13,4) (7,9, 20,9) |
YS (IRF taraf�ndan de�erlendirilen; RECIST v1.1) | n = 9 | n = 19 | n = 27 |
Olay g�r�len hastalar (%) | 3 (%33,3) | 5 (%26,3) | 8 (%29,6) |
Medyan (ay) (%95 GA) | TE (11,1, TE) | TE (TE) | TE (14,1, TE) |
PS (IRF taraf�ndan de�erlendirilen; RECIST v1.1) | n = 32 | n = 80 | n = 119 |
Olay g�r�len hastalar (%) | 24 (%75) | 59 (%73,8) | 88 (%73,9) |
Medyan (ay) (%95 GA) | 4.1 (2,3, 11,8) | 2.9 (2,1, 5,4) | 2.7 (2,1, 4,2) |
GS | n = 32 | n = 80 | n = 119 |
Olay g�r�len hastalar (%) | 18 (%56,3) | 42 (%52,5) | 59 (%49,6) |
Medyan (ay) (%95 GA) | 12,3 (6, TE) | 14,1 (9,2, TE) | 15,9 (10,4, |
|
|
| TE) |
1 y�ll�k GS oran� (%) | %52,4 | %54,8 | %57,2 |
GA = g�ven aral���; YS = yan�t s�resi; IC = t�m�r� infiltre eden imm�n h�creler; IRF = ba��ms�z de�erlendirme tesisi; TE = tahmin edilemiyor; OYO = objektif yan�t oran�; GS = genel sa�kal�m; PS = progresyonsuz sa�kal�m; RECIST = Kat� T�m�rlerde Yan�t De�erlendirme Kriterleri v1.1.
Kohort 2'de, ko-primer etkililik sonlan�m noktalar�, bir IRF taraf�ndan RECIST v1.1 kullan�larak de�erlendirilen OYO ve Modifiye RECIST (mRECIST) kriterlerine g�re ara�t�rmac� taraf�ndan de�erlendirilen OYO olarak do�rulanm��tur. 310 hasta, klinik faydan�n kaybedilmesine kadar 3 (��) haftada bir intraven�z inf�zyon yoluyla 1200 mg atezolizumab ile tedavi edilmi�tir. Kohort 2'nin primer analizi, t�m hastalar en az 24 hafta s�reyle takip edildi�inde ger�ekle�tirilmi�tir. �al��ma Kohort 2'deki ko-primer sonlan�m noktalar�n� kar��lam��; bu da �nceden belirlenmi� %10'luk bir ge�mi� kontrol yan�t oran� ile kar��la�t�r�ld���nda, IRF taraf�ndan RECIST v1.1 kullan�larak de�erlendirilen ve ara�t�rmac� taraf�ndan mRECIST kullan�larak de�erlendirilen istatistiksel olarak anlaml� OYO'lar g�stermi�tir.
Kohort 2'de 21,1 ayl�k bir medyan sa�kal�m takip s�resi ile de analiz ger�ekle�tirilmi�tir. IRF RECIST v1.1'e g�re do�rulanm�� OYO'lar, PD-L1 ekspresyonu ≥%5 olan hastalarda %28 (%95 GA: 19,5, 37,9), PD-L1 ekspresyonu ≥%1 olan hastalarda %19,3 (%95 GA: 14,2, 25,4)
ve t�m kat�lanlarda %15,8 (%95 GA: 11,9, 20,4) olmu�tur. Ara�t�rmac� taraf�ndan de�erlendirilen mRECIST'e g�re do�rulanm�� OYO ise PD-L1 ekspresyonu ≥%5 olan hastalarda %29 (%95 GA: 20,4, 38,9), PD-L1 ekspresyonu ≥%1 olan hastalarda %23,7 (%95
GA: 18,1, 30,1) ve t�m kat�lanlarda %19,7 (%95 GA: 15,4, 24,6) olmu�tur. T�m kat�lanlar pop�lasyonunda IRF RECIST v1.1'e g�re tam yan�t oran� %6,1 (%95 GA: 3,7, 9,4) olmu�tur. Kohort 2'de, herhangi bir PD-L1 ekspresyon alt grubunda veya t�m kat�lanlarda medyan YS'ye ula��lmam��; ancak PD-L1 ekspresyonu <%1 olan hastalarda ula��lm��t�r (13,3 ay; %95 GA 4,2, TE). 12. ayda GS oran� t�m kat�lanlarda %37 olmu�tur.
IMvigor130 (WO30070): Tedavi edilmemi� lokal ileri evre veya metastatik �rotelyal karsinomlu hastalarda monoterapi olarak ve platin bazl� kemoterapi ile kombinasyon halinde atezolizumab�n de�erlendirildi�i faz III �ok merkezli, randomize, plasebo kontroll� �al��ma.
Sa�kal�m verilerinin ilk a�amada g�zden ge�irilmesini takiben ba��ms�z bir Veri �zleme Kurulu (�DMC) taraf�ndan verilen tavsiyeye dayan�larak, t�m�rlerinde PD-L1 ekspresyonu d���k olan (PD-L1 ile boyanm��, t�m�r� infiltre eden imm�n h�crelerin [IC], t�m�r alan�n�n <%5'ini kaplamas�) hastalar�n atezolizumab monoterapisi koluna eklenmesi, bu alt grubun genel sa�kal�m�nda azalma g�zlemlendikten sonra durdurulmu�tur. �DMC, halihaz�rda monoterapi koluna randomize edilmi� ve monoterapi kolunda tedavi g�rmekte olan hastalar i�in herhangi bir tedavi de�i�ikli�i tavsiye etmemi�tir. Ba�ka bir de�i�iklik tavsiye edilmemi�tir.
Hepatosel�ler kanser:
IMbrave150 (YO40245): Bevacizumab ile kombinasyon halinde, daha �nce sistemik tedavi almam�� rezeke edilemeyen hepatosel�ler kanserli hastalarda randomize faz III �al��ma
Daha �nce sistemik tedavi g�rmemi� lokal ileri veya metastatik evre ve/veya rezeke edilemeyen hepatosel�ler kanserli hastalarda bevacizumab ile kombinasyon halinde atezolizumab�n
etkilili�i ve g�venlili�ini de�erlendirmek �zere, faz III, randomize, �ok merkezli, uluslararas�, a��k etiketli bir �al��ma olan IMbrave150 y�r�t�lm��t�r. Toplamda 501 hasta (2:1) oran�nda ya intraven�z inf�zyon ile her 3 haftada bir uygulanan atezolizumab (1200 mg) ve 15 mg/kg bevacizumaba ya da g�nde iki kez oral yolla uygulanan sorafenib 400 mg'a randomize edilmi�tir. Randomizasyon co�rafi b�lge (geri kalan d�nya �lkelerine kar�� Japonya hari� Asya), makrovask�ler invazyon ve/veya ekstrahepatik yay�lma (yoka kar�� var), ba�lang�� ⍺- fetoprotein (AFP) (≥400 ng/mL'ye kar�� <400 ng/mL) ve ECOG performans durumuna (1'e kar�� 0) g�re s�n�fland�r�lm��t�r. Her iki koldaki hastalar klinik fayda kayb�na veya kabul edilemez toksisiteye kadar tedavi alm��t�r. Hastalar atezolizumab veya bevacizumab� b�rak�p (�rn. advers olaylardan dolay�) klinik fayda kayb� veya tekli ajan ile ili�kili kabul edilemez toksisiteye kadar tekli ajan tedavisine devam edebilmi�tir.
�al��maya hastal��� cerrahi ve/veya b�lgesel tedavilere uygun olmayan veya cerrahi ve/veya b�lgesel tedavilerden sonra ilerleyen Child-Pugh A, ECOG 0/1'e sahip ve daha �nce sistemik tedavi almam�� yeti�kinler kaydedeilmi�tir. Kanama (�l�mc�l olaylar dahil) bevacizumab ile bilinen bir advers reaksiyondur ve �st gastrointestinal kanama HSK'li hastalarda yayg�n ve hayati risk ta��yan bir komplikasyondur. Bu nedenle, hastalar�n tedaviden �nceki 6 ay i�inde varis varl��� a��s�ndan de�erlendirilmesi zorunlu tutulmu� ve tedaviden �nceki 6 ay i�inde varise ba�l� kanama ge�irmi�lerse, kanama veya y�ksek kanama riski olan tedavi edilmemi� ya da yeterince tedavi edilmemi� varisleri varsa �al��madan hari� tutulmu�tur. Aktif hepatit B'li hastalar i�in �al��ma tedavisine ba�lamadan �nce 28 g�n i�inde HBV DNA< 500 IU/mL ve �al��maya dahil olmadan �nce ve �al��ma boyunca minimum 14 g�nl�k standart anti-HBV tedavisi gerekli olmu�tur.
Hastalar ayr�ca, orta �iddette veya �iddetli asitleri; hepatik ensefalopati �yk�leri, hepatik ensefalopati �yk�s�, bilinen fibrolameller HSKsi, sarkomatoid HSK'si, mikst kolanjiokarsinomu ve HSK'si, HBV ve HCV'nin aktif koenfeksiyonu, otoimm�n hastal�k �yk�leri varsa, randomizasyondan �nceki 4 (d�rt) hafta i�inde canl�, atten�e a�� uygulanm��sa; randomizasyondan �nceki 4 (d�rt) hafta i�inde sistemik immunostimulat�r ajan veya 2 (iki) hafta i�inde immunsupresif ila�lar uygulanm��sa; tedavi edilmemi� veya kortikosteroide ba��ml� beyin metastazlar� varsa da hari� tutulmu�tur. T�m�r de�erlendirmeleri 1. (birinci) D�ng�, 1. (birinci) G�nden sonraki ilk 54 hafta boyunca her 6 (alt�) haftada bir, ard�ndan her 9 (dokuz) haftada bir y�r�t�lm��t�r.
�al��ma pop�lasyonunun demografik ve ba�lang�� hastal�k �zellikleri tedavi kollar� aras�nda iyi dengelenmi�tir. Medyan ya� 65 olup (aral�k: 26 ila 88), hastalar�n %83'� erkektir. Hastalar�n �o�u Asyal� (%57) ve beyazd�r (%35). %40'� Asya'dan (Japonya hari�), %60'� geri kalan d�nya �lkelerindendir. Hastalar�n yakla��k %75'i makrovask�ler invazyon ve/veya esktrahepatik yay�lma ile ba�vururken, %37'sinde ba�lang�� AFP de�eri ≥400 ng/mL'dir. Ba�lang�� ECOG performans durumu 0 (%62) veya 1 (bir)'dir (%38). HSK geli�imi a��s�ndan birincil risk fakt�rleri hastalar�n %48'inde Hepatit B vir�s� enfeksiyonu, hastalar�n %22'sinde Hepatit C vir�s� enfeksiyonu ve hastalar�n %31'inde viral olmayan hastal�kt�r. HSK, hastalar�n
%82'sinde Barselona Klini�i Karaci�er Kanseri (BCLC) evre C, hastalar�n %16's�nda evre B ve hastalar�n %3 (��)'�nde evre A olarak s�n�fland�r�lm��t�r.
E�-primer etkililik sonlan�m noktalar� RECIST v1.1'e g�re GS (genel sa�kal�m) ve ba��ms�z de�erlendirme merkezi (IRF) taraf�ndan de�erlendirilen PS'dir (progresyonsuz sa�kal�m). Birincil analiz zaman�nda hastalar 8,6 ayl�k medyan sa�kal�m takibi s�resine sahiptir. Veriler sorafenibe k�yasla atezolizumab + bevacizumab ile RECIST v1.1'e g�re IRF taraf�ndan de�erlendirildi�i �zere PS ve GS'de istatistiksel anlaml� bir iyile�me g�stermi�tir. �statistiksel anlaml� bir iyile�me, ayr�ca RECIST v1.1'e ve HSK de�i�tirilmi� RECIST'e (mRECIST) g�re
IRF taraf�ndan do�rulanm�� objektif yan�t oran�nda da (OYO) g�zlenmi�tir. Primer analize ait kritik etkililik bulgular� Tablo 14'te �zetlenmektedir.
15,6 ayl�k ortanca sa�kal�m takip s�resi ile tan�mlay�c� bir g�ncellenmi� etkililik analizi y�r�t�lm��t�r. Medyan GS, atezolizumab + bevacizumab kolunda 19,2 ay (%95 GA: 17, 23,7)
iken sorafenib kolunda 13,4 ay (%95 GA: 11,4, 16,9) olup risk oran� 0,66'd�r (%95 GA: 0,52, 0,85). RECIST v1.1 do�rultusunda IRF de�erlendirmesine g�re medyan progresyonsuz sa�kal�m, atezolizumab + bevacizumab kolunda 6,9 ay (%95 GA: 5,8, 8,6) iken sorafenib
kolunda 4,3 ay (%95 GA: 4, 5,6) olup risk oran� de�eri 0,65'tir (%95 GA: 0,53, 0,81).
RECIST v1.1'e g�re IRF ile de�erlendirilen genel yan�t oran�, atezolizumab + bevacizumab kolunda %29,8 (%95 GA: 24,8, 35) iken sorafenib kolunda %11,3 (%95 GA: 6,9, 17,3) olmu�tur. Do�rulanm�� yan�t verenlerde RECIST v1.1 do�rultusunda IRF de�erlendirmesine g�re ortanca yan�t s�resi (YS), atezolizumab + bevacizumab kolunda 18,1 aya (%95 GA: 14,6, tahmin edilemiyor (TE)) k�yasla sorafenib kolunda 14,9 ay bulunmu�tur (%95 GA: 4,9, 17).
Genel sa�kal�m (g�ncellenmi� analiz) ve progresyonsuz sa�kal�m (primer analiz) i�in Kaplan- Meier e�rileri �ekil 13 ve 14'te sunulmaktad�r.
Tablo 14: Etkililik �zeti (IMbrave150 primer analiz)
Kritik etkililik sonlan�m noktas�: | Atezolizumab+bevacizumab | Sorafenib |
GS | n=336 | n=165 |
�l�m say�s� (%) | 96 (%28,6) | 65 (%39,4) |
Olaya kadar ge�en medyan s�re (ay) | TE | 13,2 |
%95 GA | (TE, TE) | (10.4, TE) |
S�n�fland�r�lm�� risk oran� | 0,58 (0,42, 0,79) | |
p de�eri | 0,0006 | |
6 ayl�k GS (%) | %84,8 | %72,3 |
IRF taraf�ndan de�erlendirilen PS, RECIST 1.1 | n=336 | n=165 |
Olay say�s� (%) | 197 (%58,6) | 109 (%66,1) |
Medyan PS s�resi (ay) | 6,8 | 4,3 |
%95 GA | (5,8, 8,3) | (4, 5,6) |
S�n�fland�r�lm�� risk oran� | 0,59 (0,47, 0,76) | |
p de�eri | <0,0001 | |
6 ayl�k PS | %54,5 | %37,2 |
IRF taraf�ndan de�erlendirilen OYO, RECIST 1.1 | n=326 | n=159 |
Do�rulanm�� yan�t veren say�s� (%) | 89 (%27,3) | 19 (%11,9) |
%95 GA | (22,5, 32,5) | (7,4, 18) |
p-de�eri | <0,0001 | |
Kritik etkililik sonlan�m noktas�: | Atezolizumab+bevacizumab | Sorafenib |
Tam yan�tlar�n say�s� (%) | 18 (%5,5) | 0 |
K�smi yan�tlar�n say�s� (%) | 71 (21,8) | 19 (%11,9) |
Stabil hastal�k say�s� (%) | 151 (%46,3) | 69 (43,4) |
IRF ile de�erlendirilen YS, RECIST 1.1 | n=89 | n=19 |
Ay cinsinden medyan | TE | 6,3 |
%95 GA | (TE, TE) | (4,7, TE) |
Aral�k (ay) | (1,3+, 13,4+) | (1,4+, 9,1+) |
IRF taraf�ndan de�erlendirilen OYO, HSK mRECIST | n=325 | n=158 |
Do�rulanm�� yan�t veren say�s� (%) | 108 (%33,2) | 21 (%13,3) |
%95 GA | (28,1, 38,6) | (8,4, 19,6) |
p-de�eri | <0,0001 | |
Tam yan�tlar�n say�s� (%) | 33 (%10,2) | 3 (%1,9) |
K�smi yan�tlar�n say�s� (%) | 75 (%23,1) | 18 (%11,4) |
Stabil hastal�k say�s� (%) | 127 (%39,1) | 66 (%41,8) |
IRF ile de�erlendirilen YS, HSK mRECIST | n=108 | n=21 |
Ay cinsinden medyan | TE | 6,3 |
%95 GA | (TE, TE) | (4,9, TE) |
Aral�k (ay) | (1,3+, 13,4+) | (1,4+, 9,1+) |
Co�rafi b�lgeye (geri kalan d�nya �lkelerine kar�� Japonya hari� Asya), makrovask�ler invazyon ve/veya ekstrahepatik yay�lma (yoka kar�� var), ba�lang�� ⍺-fetoprotein (AFP) (≥400'e kar�� <400 ng/mL) g�re s�n�fland�r�lm��t�r + Sans�rl� bir de�eri g�sterir PS = progresyonsuz sa�kal�m; RECIST = Solid T�m�rlerde Yan�t De�erlendirme Kriterleri v1.1; CC mRECIST = Hepatosel�ler Karsinom ��in De�i�tirilmi� RECIST De�erlendirmesi; GA = g�ven aral���; OYO = objektif yan�t oran�; YS = yan�t s�resi; GS = genel sa�kal�m; TE = Tahmin edilemiyor; N/A=ge�erli de�ildir | ||
�ki tarafl� s�n�fland�r�lm�� log-s�ra testi temelinde
5.2. Farmakokinetik �zellikler
Genel �zelliklerAtezolizumaba maruziyet 1 (bir) mg/kg v�cut a��rl��� - 20 mg/kg v�cut a��rl��� doz aral���nda 3 (��) haftada bir uygulanan sabit doz 1200 mg doz ile orant�l� olarak artm��t�r. 472 hastay� i�eren bir pop�lasyon analizi, a�a��daki doz aral��� i�in atezolizumab farmakokineti�ini a��klam��t�r: Birinci derece eliminasyonla bir do�rusal iki b�lmeli da��l�m modeli ile 1 - 20 mg/kg v�cut a��rl���. Bir pop�lasyon farmakokinetik analizi, 6 (alt�)-9 (dokuz) hafta tekrarl� dozlamadan sonra kararl� durumun elde edildi�ini �ne s�rmektedir. E�ri alt�ndaki alanda, maksimum konsantrasyon ve en d���k konsantrasyonda sistemik birikim s�ras�yla 1,91; 1,46 ve 2,75 kat olmu�tur.
Emilim:
Atezolizumab intraven�z inf�zyon �eklinde uygulan�r. Di�er uygulama yollar�yla yap�lan �al��malar olmam��t�r.
Da��l�m:
Pop�lasyon farmakokinetik analizi, bir hastada merkezi kompartman da��l�m hacminin 3,28 L ve kararl� durumunda hacmin 6,91 L oldu�unu g�stermektedir.
Biyotransformasyon:
Atezolizumab�n metabolizmas� do�rudan ara�t�r�lmam��t�r. Antikor klerensi esas olarak katabolizmayla ger�ekle�ir.
Eliminasyon:
Pop�lasyon farmakokinetik analizi, atezolizumab�n klerensinin 0,2 L/g�n ve tipik terminal eliminasyon yar� �mr�n�n 27 g�n oldu�unu g�stermektedir.
�zel pop�lasyonlara ili�kin ek bilgiler:
Pop�lasyon farmakokineti�i ve maruziyet-yan�t analizlerine g�re a�a��daki fakt�rlerin atezolizumab�n farmakokineti�i �zerinde bir etkisi yoktur: ya�, (21-89 ya�), b�lge, etnik k�ken, b�brek bozuklu�u, hafif karaci�er bozuklu�u, PD-L1 ekspresyonu d�zeyi veya ECOG durumu. V�cut a��rl���, cinsiyet, pozitif ADA durumu, alb�min seviyeleri ve t�m�r y�k�n�n atezolizumab farmakokineti�i �zerindeki etkisi istatistiki olarak anlaml� ancak klinik olarak
anlaml� de�ildir. Doz ayarlanmas� �nerilmemektedir. Pediyatrik pop�lasyon:
Pediyatrik (<18 ya�, n= 69) ve gen� eri�kin (18-30 ya�, n=18) hastalarda yap�lan bir erken faz, �ok merkezli, a��k etiketli �al��madan elde edilen farmakokinetik sonu�lar, atezolizumab klerensi ve da��l�m hacminin, normal v�cut a��rl���nda her 3 (��) hafta 15 mg/kg v�cut a��rl��� alan pediyatrik hastalar ve 1200 mg atezolizumab alan gen� eri�kin hastalar aras�nda kar��la�t�r�labilir oldu�unu g�stermi�tir. Maruziyetin ise, v�cut a��rl��� d��t�k�e pediyatrik hastalarda artt��� g�zlenmi�tir. Bu de�i�iklikler, atezolizumab konsantrasyonunun terap�tik hedef maruziyetinin alt�na d��mesi ile ba�lant�l� de�ildir. 2 ya� alt�ndaki �ocuklar i�in veriler s�n�rl�d�r, bu nedenle kesin sonu�lara var�lamamaktad�r.
Geriyatrik pop�lasyon:
Ya�l� hastalarda TECENTRIQ i�in �zel bir �al��ma yap�lmam��t�r. Ya��n atezolizumab�n farmakokineti�i �zerindeki etkisi bir pop�lasyon farmakokinetik analizinde de�erlendirilmi�tir. Ya�, 21-89 ya� aral���ndaki (n=472) ve medyan 62 ya��ndaki hastalar temel al�nd���nda, atezolizumab�n farmakokineti�ini etkileyen �nemli bir kovaryant olarak tan�mlanmam��t�r. <65 ya��ndaki (n=274), 65-75 ya��ndaki (n=152) ve >75 ya��ndaki (n=46) hastalarda atezolizumab�n farmakokineti�inde klinik a��dan anlaml� bir fark g�zlenmemi�tir (bkz. B�l�m 4.2).
B�brek yetmezli�i:
B�brek bozuklu�u olan hastalarda TECENTRIQ i�in �zel bir �al��ma yap�lmam��t�r. Pop�lasyon farmakokinetik analizinde, b�brek fonksiyonu normal (90 mL/dk/1,73 m veya �zeri eGFR; n=140) olan hastalarla kar��la�t�r�ld���nda, hafif (60 - 89 mL/dk/1,73 m eGFR; n=208) veya orta �iddetli (30 - 59 mL/dk/1,73 m eGFR; n=116) b�brek bozuklu�u olan hastalarda atezolizumab�n klerensinde klinik a��dan �nemli farklar bulunmam��t�r. Yaln�zca birka� hastada �iddetli b�brek bozuklu�u vard�r (eGFR 15 - 29 mL/dk/1,73 m; n=8) (bkz. B�l�m 4.2). �iddetli b�brek yetmezli�inin atezolizumab�n farmakokineti�i �zerindeki etkisi bilinmemektedir.
Karaci�er yetmezli�i:
Karaci�er bozuklu�u olan hastalarda TECENTRIQ i�in �zel bir �al��ma yap�lmam��t�r. Pop�lasyon farmakokinetik analizinde hafif karaci�er bozuklu�u (bilirubin ≤N�S ve AST>N�S veya bilirubin <1 – 1,5 x N�S ve herhangi bir AST) veya orta d�zeyde karaci�er bozuklu�u (bilirubin <1,5 – 3 x N�S ve herhangi bir AST) olan hastalarda normal karaci�er fonksiyonu (bilirubin ve AST ≤N�S) olan hastalara k�yasla atezolizumab�n klerensi bak�m�ndan klinik olarak �nemli farklar bulunmam��t�r. �iddetli karaci�er bozuklu�u olan hastalara ili�kin veri mevcut de�ildir (bilirubin > 3 x N�S ve herhangi bir AST). Karaci�er bozuklu�u, Ulusal Kanser Enstit�s� (NCI) karaci�er fonksiyon bozuklu�u kriterlerine g�re tan�mlanm��t�r (bkz. B�l�m 4.2). �iddetli karaci�er yetmezli�inin (bilirubin > 3 x N�S ve herhangi bir AST) atezolizumab�n farmakokineti�i �zerindeki etkisi bilinmemektedir.
5.3. Klinik �ncesi g�venlilik verileri
Karsinojenisite:TECENTRIQ'in karsinojenik potansiyelini belirlemek i�in karsinojenisite �al��mas� yap�lmam��t�r.
Mutajenisite:
TECENTRIQ'in mutajenik potansiyelini belirlemek i�in mutajenisite �al��mas� yap�lmam��t�r. Bununla birlikte, monoklonal antikorlar�n DNA veya kromozomlar� de�i�tirmesi beklenmemektedir.
Fertilite:
TECENTRIQ ile herhangi bir fertilite �al��mas� yap�lmam��t�r. Bununla birlikte, kronik toksisite �al��mas�na sinomolgus maymunlar�nda erkek ve di�i �reme organlar�n�n de�erlendirilmesi dahil edilmi�tir. Atezolizumab�n di�i maymunlara tahmini EAA'da uygulanmas� (�nerilen dozu alan hastalardaki EAA'n�n yakla��k 6 kat�), geri d�n���ml� olarak d�zensiz adet d�ng�s� paternine ve yumurtal�klarda yeni olu�turulmu� korpus lutea eksikli�ine neden olmu�tur. Erkek �reme organlar� �zerinde herhangi bir etki olmam��t�r.
Teratojenisite:
TECENTRIQ ile hayvanlarda �reme veya teratojenisite �al��malar� yap�lmam��t�r. Hayvan �al��malar�, PD-L1/PD-1 yola��n�n inhibisyonunun b�y�mekte olan fet�s�n reddi riskinde imm�nite ile ili�kili art��a ve b�ylelikle fetal �l�me yol a�abilece�ini ortaya koymu�tur. TECENTRIQ uygulamas�n�n gebelik �zerinde advers bir reaksiyona yol a�mas� beklenir ve embriyo-fetal letalitesi de dahil olmak �zere insan fet�s� i�in bir risk olu�turur.
6. FARMAS�T�K �ZELL�KLER
6.1. Yard�mc� maddelerin listesi
L-histidin
Glasiyal asetik asit S�kroz
Polisorbat 20 Enjeksiyonluk su
6.2. Ge�imsizlikler
6.2. Ge�imsizlikler
6.3. Raf �mr�
A��lmam�� flakon: 36 ay
Seyreltilmi� ��zelti: Haz�rlama zaman�ndan sonra ortam s�cakl���nda (≤30°C) 24 saat ve 2- 8°C'de 30 g�ne kadar kullan�mdaki kimyasal ve fiziksel stabilite g�sterilmi�tir.
Mikrobiyolojik a��dan, haz�rlanan inf�zyonluk ��zelti hemen kullan�lmal�d�r. Hemen kullan�lmazsa, kullan�m s�ras�ndaki saklama s�releri ve kullan�m �ncesi ko�ullar kullan�c�n�n sorumlulu�undad�r ve normalde, seyreltme kontroll� ve valide aseptik ko�ullarda yap�lmad��� s�rece, 2-8°C'de 24 saatten fazla veya ortam s�cakl���nda (≤25°C) 8 saatten fazla olmamal�d�r.
6.4. Saklamaya y�nelik �zel tedbirler
Buzdolab�nda saklay�n�z (2°C-8°C).
I��ktan korumak i�in flakonu karton kutusunda saklay�n�z. Dondurmay�n�z. �alkalamay�n�z.
T�bbi �r�n�n seyreltme sonras�nda saklama ko�ullar� i�in bkz. B�l�m 6.3.
6.5. Ambalaj�n niteli�i ve i�eri�i
20 mL inf�zyonluk konsantre ��zelti i�eren tapal� (butil kau�uk) flakon (Tip I cam). Bir flakonluk paket.
6.6. Be�eri t�bbi �r�nden arta kalan maddelerin imhas� ve di�er �zel �nlemler
TECENTRIQ herhangi bir antimikrobiyal koruyucu veya bakteriyostatik ajan i�ermemektedir ve haz�rlanan ��zeltinin steril olmas�n� sa�lamak i�in bir sa�l�k mesle�i mensubu taraf�ndan aseptik teknik ile haz�rlanmal�d�r.
Aseptik haz�rlama, ta��ma ve saklama
�nf�zyonun haz�rlanmas� s�ras�nda aseptik temas sa�lanmal�d�r. Haz�rlama i�lemi:
E�itilmi� personel taraf�ndan aseptik ko�ullar alt�nda, �zellikle de parenteral �r�nlerin aseptik haz�rlanmas�na ili�kin iyi uygulama kurallar� do�rultusunda yap�lmal�d�r.
�ntraven�z ajanlar�n g�venli temas� i�in standart �nlemleri kullanarak laminer hava ak�m� veya biyolojik g�venlik kabininde haz�rlanmal�d�r.
Aseptik ko�ullar�n idamesini sa�lamak i�in haz�rlanan intraven�z inf�zyon ��zeltisine uygun saklama i�lemi ile devam etmelidir.
�alkalamay�n�z.
Seyreltme talimatlar�:
20 mL TECENTRIQ konsantresi flakondan �ekilmeli ve i�inde sodyum klor�r 9 mg/mL (% 0,9) enjeksiyonluk ��zelti bulunan bir PVC, polietilen (PE) veya poliolefin inf�zyon torbas� i�ine seyreltilmelidir. Seyreltmeden sonra ��zeltinin final konsantrasyonu 3,2 ila 16,8 mg/mL olmal�d�r.
Torba, k�p�k olu�umuna izin vermeden ��zeltiyi kar��t�rmak i�in yava��a alt �st edilmelidir. �nf�zyon haz�rland���nda hemen uygulanmal�d�r (bkz. B�l�m 6.3).
Parenteral t�bbi �r�nler uygulanmadan �nce partik�ller ve renk de�i�imi a��s�ndan ��plak g�zle
incelenmelidir. Partik�ller veya renk de�i�imi g�zlenirse ��zelti kullan�lmamal�d�r.
TECENTRIQ ile �r�ne temas eden polivinil klor�r (PVC), polietilen (PE) veya poliolefin (PO) y�zeyleri olan intraven�z torbalar aras�nda ge�imsizlik g�zlenmemi�tir. �lave olarak, polieters�lfon veya polis�lfon i�eren d�z eksenli filtre membranlar� ve inf�zyon setleri ile PVC, PE, polibutadien veya polieter�retan i�eren di�er inf�zyon yard�mc�lar� ile de ge�imsizlik g�zlenmemi�tir. D�z eksenli filtre membranlar�n�n kullan�lmas� se�ime ba�l�d�r.
Di�er ila�lar� ayn� inf�zyon hatt�nda birlikte uygulamay�n�z.
Kullan�lmam��/son kullanma tarihi ge�mi� ila�lar�n imhas�:
T�bbi �r�nlerinin �evreye sal�nmas� en aza indirilmelidir. �la�lar, at�k suyla birlikte at�lmamal�d�r ve evsel at�klarla imhas�ndan ka��n�lmal�d�r.
Kullan�lmam�� olan �r�nler ya da at�k materyaller, “T�bbi At�klar�n Kontrol� Y�netmeli�i'' ve “Ambalaj At�klar�n�n Kontrol� Y�netmeli�i”ne uygun olarak imha edilmelidir.
 Depresyonu Anlamak
Depresyon farkl� ki�ileri farkl� bi�imlerde etkiler. Duygusal veya fiziksel
olmak �zere geni� alanda belirtilere sebep olabilir.Depresyona neler sebep olur?
Depresyonu Anlamak
Depresyon farkl� ki�ileri farkl� bi�imlerde etkiler. Duygusal veya fiziksel
olmak �zere geni� alanda belirtilere sebep olabilir.Depresyona neler sebep olur? |
 Artrit
Artrit, olduk�a yayg�n bir hastal�kt�r ancak iyi anla��lamam��t�r. Asl�nda �artrit� tek bir hastal���n ad� de�ildir; eklem a�r�s� veya eklem hastal�klar�n� adland�rman�n gayri resmi yoludur.
Artrit
Artrit, olduk�a yayg�n bir hastal�kt�r ancak iyi anla��lamam��t�r. Asl�nda �artrit� tek bir hastal���n ad� de�ildir; eklem a�r�s� veya eklem hastal�klar�n� adland�rman�n gayri resmi yoludur. |
 |
�izofrenlik �izofrenli�in psikiatrik te�hisi hakk�nda �ok fazla anla�mazl�k vard�r. Bu sayfadaki bilgiler, �izofrenli�in te�hisi, nedenleri ve tedavisi hakk�ndaki fakl� teoriler hakk�nda bilgi verecektir. |
 |
Travma Sonras� Bunal�m� Travmatik bir olay, g�nl�k ola�an olaylar�n d���nda olan ve ki�iyi derinden rahats�z eden bir olayd�r.Bir�ok olay b�yle bir etki g�sterebilir. |
 |
Deri Kanseri Deri kanseri �ok rastlanan bir hastal�kt�r. �� ana t�r� bulunur ;genelde kemirici �lser olarak bilinen bazal h�creli karsinom, yass� h�creli karsinom ve k�t� huylu t�m�r. |
�LA� GENEL B�LG�LER�
Roche M�stahzarlar� Sanayi A.�.
| Sat�� Fiyat� | 123966.5 TL [ 3 Aug 2026 ] |
| �nceki Sat�� Fiyat� | 123966.5 TL [ 27 Jul 2026 ] |
| Original / Jenerik | Original �la� |
| Re�ete Durumu | Normal Re�eteli bir ila�d�r. |
| Barkodu | 8699505763460 |
| Etkin Madde | Atezolizumab |
| ATC Kodu | L01XC32 |
| Birim Miktar | 1200/20 |
| Birim Cinsi | MG/ML |
| Ambalaj Miktar� | 1 |
| Antineoplastik ve �mm�nomod�lat�r Ajanlar > Di�er Kanser �la�lar� |
| �thal ( ref. �lke : Isvicre ) ve Be�eri bir ila�d�r. |