SOFEXAN 200 mg film kapl� tablet K�sa �r�n Bilgisi
{ Sorafenib }
1. BE�ER� TIBB� �R�N�N ADI
SOFEXAN 200 mg film kapl� tablet
2. KAL�TAT�F VE KANT�TAT�F B�LE��M
Etkin madde
Sorafenib 200 mg (274,09 mg sorafenib tosilat olarak)
Yard�mc� maddeler
Yard�mc� maddeler i�in 6.1'e bak�n�z.
3. FARMAS�T�K FORMU
Film kapl� tablet
Koyu pembe renkli, yuvarlak, bombeli film kapl� tabletler
4. KL�N�K �ZELL�KLER
4.1. Terap�tik endikasyonlar
SOFEXAN;
Metastatik renal h�creli karsinoma (mRHK) tedavisinde interferon alfa ve/veya interl�kin-2 ile yan�t al�namad��� veya bu tip tedavilerin yan etki nedeniyle uygun olmad��� durumlarda kullan�lmas� i�in endikedir.
4.2. Pozoloji ve uygulama �ekli
Eri�kinlerPozoloji:
�nerilen SOFEXAN dozu g�nde toplam 800 mg'd�r.
Uygulama s�kl��� ve s�resi:
�nerilen toplam g�nl�k SOFEXAN dozu, g�nde iki kez 400 mg (2 x 2 tablet) tablet �eklinde al�n�r.
Tedavi, hasta art�k tedaviden daha fazla klinik yarar g�rmeyinceye veya kabul edilemez �l��de yan etki ortaya ��k�ncaya kadar s�rd�r�lmelidir.
Uygulama �ekli:
Antikanser tedavide deneyimli bir hekim kontrol�nde kullan�lmal�d�r.
SOFEXAN oral uygulama i�indir ve bir bardak su ile yutulmal�d�r. Tabletler a� kar�na ya da d���k veya orta ya�l� bir ���n ile birlikte al�nabilir. Tabletler y�ksek ya�l� bir ���nle al�nacaksa, yemekten en az 1 saat �nce veya yemekten 2 saat sonra al�nmal�d�r.
Doz titrasyonu, doz ayarlamas�, �zel izleme �nerileri:
Hepatosel�ler Karsinoma (HSK) ve �lerlemi� Renal H�creli Karsinomada (RHK) Doz Azalt�m�
Ku�kulu advers ila� reaksiyonlar�n�n tedavisinde, sorafenib tedavisinin ge�ici olarak durdurulmas� ve/veya dozun azalt�lmas� gerekli olabilir.
HSK ve ilerlemi� RHK tedavisinde doz azalt�m� gerekli oldu�unda, sorafenib dozu g�nde bir kez 400 mg'a (1 x 2 tablet) azalt�lmal�d�r (bkz. B�l�m 4.4).
Deri toksisitesi durumunda �nerilen doz modifikasyonlar� Tablo 1'de �zetlenmi�tir.
Tablo 1: HSK ve RHK ile Deri Toksisitesi Durumunda �nerilen Doz Modifikasyonlar�
Derece | Ortaya ��k�� | SOFEXAN doz modifikasyonu |
Derece 1: El veya ayaklarda hastan�n normal aktivitelerini etkilemeyen uyu�ma, disestezi, parestezi, kar�ncalanma, a�r�s�z rahats�zl�k | Herhangi bir zaman | Derhal destekleyici �nlemler al�n�r ve SOFEXAN tedavisine devam edilir. |
Derece 2: El veya ayaklarda hastan�n normal aktivitelerini etkileyen a�r�l� eritem ve �i�me ve/veya rahats�zl�k | �lk olay | Derhal destekleyici �nlemler al�n�r ve SOFEXAN dozu 28 g�n s�re ile g�nde 400 mg'a d���r�l�r. Doz azalt�m�ndan sonra toksisite derece 0-1'e gerilerse, 28 g�n sonra sorafenib dozu art�r�larak tam doza ge�ilir. Doz azalt�m�na kar��n toksisite derece 0-1'e gerilemezse, en az 7 g�n s�reyle, toksisite derece 0- 1'e d�n�nceye kadar SOFEXAN tedavisine ara verilir. Aradan sonra tedaviye yeniden ba�larken, SOFEXAN 28 g�n s�reyle g�nde bir kez 400 mg'l�k azalt�lm�� doz �eklinde uygulan�r. Doz azalt�m�yla toksisite derece 0-1'de tutulabilirse, 28 g�n sonra SOFEXAN dozu art�r�larak tam doza ge�ilir. |
| 7 g�n i�inde iyile�me yoksa veya 2. veya 3. olu�um | Toksisite derece 0-1'e gerileyene dek SOFEXAN tedavisine ara veriniz. Tedaviye yeniden ba�larken, SOFEXAN dozunu bir doz d�zeyi azalt�n�z (g�nde 400 mg veya iki g�nde bir 400 mg). |
D�rd�nc� kez | SOFEXAN tedavisini sonland�r�n�z. | |
Derece 3: El veya ayaklarda hastan�n �al��mas�n� ve g�nl�k aktivitelerini ger�ekle�tirmesini engelleyen nemli deskuamasyon, �lserasyon, kabarma veya �iddetli a�r� veya �iddetli rahats�zl�k | �lk olay | Derhal destekleyici �nlemler al�n�r ve en az 7 g�n s�reyle, toksisite derece 0-1'e gerileyinceye kadar SOFEXAN tedavisine ara verilir. Aradan sonra tedaviye yeniden ba�larken, SOFEXAN 28 g�n s�reyle g�nde bir kez 400 mg'l�k azalt�lm�� doz �eklinde uygulan�r. Doz azalt�m�yla toksisite derece 0-1'de tutulabilirse, 28 g�n sonra SOFEXAN dozu art�r�larak tam doza ge�ilir. |
�kinci kez | �lk ortaya ��k��taki gibi davran�l�r, ancak SOFEXAN tedavisi yeniden ba�lat�ld�ktan sonra, s�rekli olarak azalt�lm�� g�nl�k 400 mg dozu uygulan�r. | |
���nc� kez | SOFEXAN tedavisini sonland�r�n�z. |
Diferansiye Tiroid Karsinomada (DTK) Doz Azalt�m�
Ku�kulu advers ila� reaksiyonlar�n�n tedavisinde, sorafenib tedavisinin ge�ici olarak durdurulmas� ve/veya dozun azalt�lmas� gerekli olabilir.
DTK tedavisinde doz azalt�m� gerekli oldu�unda, sorafenib dozu b�l�nm�� dozlar �eklinde g�nl�k 600 mg'a (12 saat arayla bir seferde 200 mg'l�k iki tablet ve di�er seferde 200 mg'l�k bir tablet olacak �ekilde g�nde toplam 3 tablet) azalt�lmal�d�r.
Ek doz azalt�m� gerekli olursa, sorafenib dozu g�nde iki kez 200 mg'l�k bir tablete, ard�ndan g�nde bir kez 200 mg'l�k bir tablete indirilebilir. Hematolojik olmayan advers reaksiyonlar�n iyile�mesinden sonra, sorafenib dozu art�r�labilir.
Tablo 2: Diferansiye Tiroid Kanserli Hastalarda �nerilen Doz Azalt�m� D�zeyleri
Doz d�zeyi | SOFEXAN dozu |
|
0 | 800 mg g�nl�k doz | (G�nde iki kez 400 mg, g�nde iki kez 2 tablet) |
-1 | 600 mg g�nl�k doz | (12 saat arayla 400 mg ve 200 mg, 12 saat arayla 2 tablet ve 1 tablet-herhangi bir doz �nce al�nabilir) |
-2 | 400 mg g�nl�k doz | (G�nde iki kez 200 mg, g�nde iki kez 1 tablet) |
-3 | 200 mg g�nl�k doz | (G�nde bir kez bir 200 mg, g�nde bir kez 1 tablet) |
Tablo 3: Diferansiye Tiroid Kanserli Hastalarda Deri Toksisitesi Durumunda �nerilen Doz Modifikasyonlar�
Derece | Ortaya ��k�� | SOFEXAN doz modifikasyonu* |
Derece 1 | Herhangi bir zaman | Derhal destekleyici �nlemler al�n�r ve SOFEXAN tedavisine devam edilir. |
Derece 2 | �lk olay | Derhal destekleyici �nlemler al�n�r ve SOFEXAN dozu g�nde 600 mg'a d���r�l�r (12 saat arayla 400 mg ve 200 mg). E�er 7 g�n i�inde iyile�me g�r�lmezse, tablonun devam�n� inceleyiniz. |
7 g�n i�inde iyile�mezse veya ikinci kez ortaya ��karsa | Derece 0-1'e gerileyinceye kadar, SOFEXAN'a ara verilir. SOFEXAN yeniden ba�lat�l�rken, doz bir doz d�zeyi azalt�l�r. | |
���nc� kez | Derece 0-1'e gerileyinceye kadar, SOFEXAN'a ara verilir. SOFEXAN yeniden ba�lat�l�rken, doz iki doz d�zeyi azalt�l�r. | |
D�rd�nc� kez | SOFEXAN tedavisi tamamen sonland�r�l�r. | |
Derece 3 | �lk olay | Derece 0-1'e gerileyinceye kadar, SOFEXAN'a ara verilir. SOFEXAN yeniden ba�lat�l�rken, doz bir doz d�zeyi azalt�l�r. |
�kinci kez | Derece 0-1'e gerileyinceye kadar, SOFEXAN'a ara verilir. SOFEXAN yeniden ba�lat�l�rken, doz iki doz d�zeyi azalt�l�r. | |
���nc� kez | SOFEXAN tedavisi tamamen sonland�r�l�r. |
* Derece 2 veya 3 deri toksisitesi i�in doz azalt�m� gereken hastalarda, azalt�lm�� SOFEXAN dozu ile en az 28 g�n tedaviden sonra deri toksisitesi derece 0-1'e gerilerse, SOFEXAN dozu art�r�labilir.
�zel pop�lasyonlara ili�kin ek bilgiler:
B�brek/ karaci�er yetmezli�i:
Hafif, orta dereceli ya da diyaliz gerektirmeyen �iddetli b�brek yetmezli�i olan hastalarda doz ayarlamas� gerekli de�ildir. Sorafenib diyalize girmekte olan hastalarda incelenmemi�tir (bkz. B�l�m 5.2).
B�brek fonksiyon bozuklu�u riski olan hastalarda s�v� dengesinin ve elektrolitlerin izlenmesi �nerilir.
Child-Pugh A veya B karaci�er yetmezli�i olan hastalarda doz ayarlamas� gerekli de�ildir. Sorafenib, Child-Pugh C karaci�er yetmezli�i olan hastalarda incelenmemi�tir (bkz. B�l�m 5.2).
Pediyatrik pop�lasyon:
Sorafenibin �ocuklardaki ve 18 ya� alt�ndaki ad�lesanlarda g�venlili�i ve etkilili�i belirlenmemi�tir.
Geriyatrik pop�lasyon:
Ya�l� hastalarda (65 ya� �zeri) doz ayarlamas� gerekli de�ildir.
Di�er:
Hastan�n cinsiyeti ya da v�cut a��rl��� temelinde doz ayarlamas� gerekli de�ildir.
4.3. Kontrendikasyonlar
SOFEXAN, sorafenibe ya da SOFEXAN'�n di�er bile�enlerinden (bkz. B�l�m 6.1) herhangi birine kar�� �iddetli a��r� duyarl�l��� bilinen hastalarda kontrendikedir.
4.4. �zel kullan�m uyar�lar� ve �nlemleri
Anevrizmalar ve arter diseksiyonlar�:
VEGF yolak inhibit�rlerinin, hipertansiyonu olan veya olmayan hastalarda kullan�lmas�, anevrizmalar ve/veya arter diseksiyonlar� olu�umunu kolayla�t�rabilir. SOFEXAN ba�lamadan �nce hipertansiyon veya anevrizma �yk�s� gibi risk fakt�rleri olan hastalarda bu risk dikkatle de�erlendirilmelidir.
Dermatolojik toksisiteler:
El-ayak deri reaksiyonu (palmar-plantar eritrodisestezi) ve d�k�nt� sorafenibe ba�l� en yayg�n advers ila� reaksiyonlar�n� temsil etmektedir. D�k�nt� ve el-ayak deri reaksiyonu s�kl�kla CTC (National Cancer Institute Common Toxicity Criteria) derece 1 ve 2'dir ve genel olarak sorafenib tedavisinin ilk alt� haftas� i�inde ortaya ��kmaktad�r.
Dermatolojik toksisitelerin tedavisinde semptomatik iyile�me ama�l� topikal tedaviler, tedavinin ge�ici olarak durdurulmas� ve/veya sorafenib doz modifikasyonu ya da �iddetli veya �srarl� durumlarda sorafenib tedavisine tamamen son verilmesi gibi y�ntemler uygulanabilir (bkz. B�l�m 4.8).
Hipertansiyon riski:
Sorafenib tedavisi alan hastalarda hipertansiyon insidans�nda art�� g�zlenmi�tir.
SOFEXAN uygulanan ilk 6 hafta boyunca kan bas�nc� haftal�k olarak takip edilmelidir. Bu s�reden sonra, kan bas�nc� izlenmeli ve hipertansiyon, gerekirse, standart t�bbi uygulama do�rultusunda tedavi edilmelidir. HSK �al��mas�nda, hipertansiyon sorafenib ile tedavi edilen hastalar�n yakla��k %9,4'�nde ve plasebo ile tedavi edilen gruptaki hastalar�n %4,3'�nde bildirilmi�tir. RHK �al��mas� 1'de, hipertansiyon sorafenib ile tedavi edilen hastalar�n yakla��k %16,9'unda ve plasebo ile tedavi edilen gruptaki hastalar�n %1,8'inde bildirilmi�tir. DTK �al��mas�nda, hipertansiyon sorafenib ile tedavi edilen hastalar�n yakla��k %40,6's�nda ve plasebo ile tedavi edilen hastalar�n %12,4'�nde bildirilmi�tir. Hipertansiyon s�kl�kla hafif ile orta derecelidir, tedavinin erken d�nemlerinde ortaya ��kar ve standart antihipertansif rejimlerle tedavi edilebilir niteliktedir. Kan bas�nc� d�zenli olarak izlenmeli ve gerekli oldu�unda standart t�bbi uygulama do�rultusunda tedavi edilmelidir. Uygun ve yeterli bir antihipertansif tedaviye kar��n �iddetli ya da �srarl� hipertansiyon veya hipertansif kriz olgular�nda, sorafenibin tamamen kesilmesi g�ndeme getirilmelidir (bkz. B�l�m 4.8). HSK �al��mas�nda sorafenib ile tedavi edilen 297 hastadan 1'inde, RHK �al��mas� 1'de sorafenib ile tedavi edilen 451 hastadan 1'inde ve DTK �al��mas�nda sorafenib ile tedavi edilen 207 hastadan 1'inde hipertansiyon nedeniyle tedavinin kal�c� olarak kesilmesi gerekmi�tir.
Hipoglisemi:
Sorafenib tedavisi s�ras�nda, klinik a��dan semptomatik ve bilin� kayb� nedeniyle hastaneye yat�� gerektiren baz� olgular dahil olmak �zere, kan glukoz de�erinde azalmalar bildirilmi�tir. Semptomatik hipoglisemi geli�ti�i takdirde, sorafenib ge�ici olarak kesilmelidir. Diyabetik hastalardaki anti-diyabetik t�bbi �r�n dozaj�n�n ayarlanmas�n�n gerekip gerekmedi�inin de�erlendirilmesi i�in kan glukoz d�zeyleri d�zenli olarak kontrol edilmelidir.
Hemoraji riski:
Sorafenib uygulamas�n� takiben kanama riskinde art�� ortaya ��kabilir. �iddetli kanama olaylar�n�n insidans� seyrektir. HSK �al��mas�nda, nedensellikten ba��ms�z olarak a��r� kanama belirgin olarak izlenmemi� ve �zofagus varislerinde kanama oran� sorafenib ile tedavi edilen hastalarda %2,4 ve plasebo ile tedavi edilen hastalarda %4 olarak kaydedilmi�tir. Herhangi bir b�lgede �l�mle sonu�lanan kanama, sorafenib ile tedavi edilen hastalar�n
%2,4'�nde ve plasebo ile tedavi edilen hastalar�n %4'�nde bildirilmi�tir. RHK �al��mas� 1'de, nedensellikten ba��ms�z olarak kanama sorafenib ile tedavi edilen gruptaki hastalar�n
%15,3'�nde ve plasebo ile tedavi edilen gruptaki hastalar�n %8,2'sinde bildirilmi�tir. CTCAE Derece 3 ve 4 kanama, sorafenib ile tedavi edilen hastalarda s�ras�yla %2 ve %0 olarak kaydedilirken, plasebo ile tedavi edilen hastalarda s�ras�yla %1,3 ve %0,2 olarak bildirilmi�tir. RHK �al��mas�nda her tedavi grubunda bir �l�mc�l hemoraji ger�ekle�mi�tir. DTK �al��mas�nda, kanama sorafenib ile tedavi edilen hastalar�n %17,4'�nde ve plasebo ile tedavi edilen hastalar�n %9,6's�nda bildirilmi� ancak CTCAE Derece 3 kanama insidans� sorafenib ile tedavi edilen hastalarda %1 ve plasebo ile tedavi edilen hastalarda %1,4 olmu�tur. Derece 4 kanama hi� bildirilmezken, plasebo edilen bir hastada �l�mc�l hemoraji meydana gelmi�tir. E�er herhangi bir kanama olay� t�bbi giri�im gerektirirse sorafenib tedavisinin tamamen kesilmesi g�ndeme getirilmelidir (bkz. B�l�m 4.8). Potansiyel kanama riski nedeniyle diferansiye tiroid kanserli hastalarda sorafenib uygulanmadan �nce trakeal, bron�iyal ve �zefageal infiltrasyon lokalize terapi ile tedavi edilmelidir.
Varfarin:
Sorafenib tedavisinde iken varfarin alan baz� hastalarda, yayg�n olmayan kanama olaylar� ya da Uluslararas� Normalize Oran (INR) de�erlerinde y�kselmeler bildirilmi�tir. E� zamanl� olarak varfarin almakta olan hastalar, protrombin zaman�nda uzama, INR ve klinik kanama epizodlar� i�in d�zenli �ekilde izlenmelidir (bkz. B�l�m 4.8).
Yara iyile�mesi komplikasyonlar�:
Sorafenibin yara iyile�mesi �zerindeki etkisi konusunda randomize bir �al��ma y�r�t�lmemi�tir. Maj�r cerrahi giri�im ge�irecek hastalarda bir �nlem olarak sorafenib tedavisinin ge�ici olarak durdurulmas� �nerilir. Maj�r cerrahi giri�im sonras�nda tedaviye yeniden ba�lama zaman� konusundaki klinik deneyim k�s�tl�d�r. Bu nedenle, maj�r cerrahi giri�im sonras�nda sorafenib tedavisine tekrar ba�lama karar� yara iyile�mesinin yeterlili�ine y�nelik klinik yarg�ya dayand�r�lmal�d�r.
Ya�l� pop�lasyon:
B�brek yetmezli�i olgular� bildirilmi�tir. B�brek fonksiyonunun izlenmesi d���n�lmelidir.
QT aral���n�n uzamas�:
Sorafenibin ventrik�ler aritmiler a��s�ndan riskin artmas�na neden olabilecek bir durum olan QT/QTc aral���n� uzatt��� g�sterilmi�tir (bkz. B�l�m 5.1). Konjenital uzun QT sendromu olan hastalarda SOFEXAN kullan�m�ndan ka��n�n�z. Sorafenib, y�ksek k�m�latif dozda antrasiklin tedavisi g�ren, baz� antiaritmik ila�lar� veya QT uzamas�na neden olan di�er t�bbi
�r�nleri kullanan hastalar gibi QTc uzamas� olan veya geli�ebilecek hastalar ile hipokalemi, hipokalsemi veya hipomagnezemi gibi elektrolit bozukluklar� olan hastalarda dikkatli kullan�lmal�d�r. Bu hastalarda SOFEXAN kullan�m� s�ras�nda elektrokardiyogram ve elektrolit �l��m� (magnezyum, kalsiyum, potasyum) ile periyodik izlem g�z �n�nde bulundurulmal�d�r. QTc aral��� 500 milisaniyenin �zerinde oldu�u veya ba�lang�ca g�re 60 milisaniye veya �zeri art�� g�sterdi�i takdirde SOFEXAN kullan�m� durdurulmal�d�r.
Kardiyak iskemi ve/veya enfarkt�s:
Randomize, plasebo kontroll�, �ift k�r bir �al��mada (�al��ma 1, bkz. B�l�m 5.1), tedaviye ba�l� kardiyak iskemi/enfarkt�s olaylar� sorafenib grubunda (%4,9), plasebo grubuna (%0,4) k�yasla daha y�ksektir. �al��ma 3'te (bkz. B�l�m 5.1), tedaviye ba�l� kardiyak iskemi/enfarkt�s olaylar�n�n insidans� sorafenib grubunda %2,7 iken, plasebo grubunda
%1,3't�r. Stabil olmayan koroner arter hastal��� �yk�s� olan ya da yak�n zamanda miyokard enfarkt�s� ge�iren hastalar bu �al��man�n d���nda b�rak�lm��t�r. Kardiyak iskemi ve/veya enfarkt�s ge�iren hastalarda sorafenibe ge�ici olarak ya da tamamen son verilmesi g�ndeme getirilmelidir (bkz. B�l�m 4.8, B�l�m 5.1).
Gastrointestinal perforasyon:
Gastrointestinal perforasyon, sorafenib almakta olan hastalarda seyrek g�r�len bir advers olayd�r. Hastalar�n %1'inden daha az�nda bildirilmi�tir. Olgular�n baz�lar�nda bu durum, g�r�n�r bir intraabdominal t�m�r ile ili�kilendirilememi�tir. Gastrointestinal perforasyon durumunda sorafenib tedavisine son verilmelidir (bkz. B�l�m 4.8).
T�m�r lizis sendromu (TLS):
Sorafenib ile tedavi edilen hastalarda pazarlama sonras� g�zetimde baz�lar� �l�mc�l olan t�m�r lizis sendromu (TLS) vakalar� bildirilmi�tir. TLS i�in risk fakt�rleri aras�nda y�ksek t�m�r y�k�, �nceden var olan kronik b�brek yetmezli�i, olig�ri, dehidratasyon, hipotansiyon ve asidik idrar bulunur. Klinik olarak tespit edildi�inde, bu hastalar yak�ndan izlenmeli, derhal tedavi edilmeli ve profilaktik hidrasyon d���n�lmelidir.
Karaci�er yetmezli�i:
Child-Pugh C (�iddetli) karaci�er yetmezli�i olan hastalara ili�kin veri bulunmamaktad�r. Sorafenib ba�l�ca karaci�er yoluyla elimine edildi�i i�in �iddetli karaci�er yetmezli�i olan hastalarda ilaca sistemik maruziyet artabilir (bkz. B�l�m 5.2).
�la� ind�kl� hepatit:
Sorafenib ind�kl� hepatit, karaci�er yetmezli�i ve �l�mle sonu�lanabilecek �ekilde, transaminazlarda anlaml� art��larla birlikte seyreden hepatosell�ler paternde karaci�er hasar�yla karakterizedir. Bilirubin ve INR'de de art�� g�r�lebilir. Global monoterapi veritaban�nda, normalin �st s�n�r�n�n 20 kat �zerine ��kan transaminaz d�zeyleri veya anlaml� klinik sekel b�rakan transaminaz art��lar� (�rne�in INR art���, assit, �l�mc�l sonu� veya transplantasyon) olarak tan�mlanan �iddetli ila� ind�kl� karaci�er hasar� insidans� 3.357 hastada iki hasta (%0,06) olarak kay�tl�d�r. Karaci�er fonksiyon testleri d�zenli olarak izlenmelidir. Viral hepatit veya altta yatan malignitede progresyon gibi alternatif bir a��klamas� olmayan anlaml� transaminaz art��� izlendi�i takdirde SOFEXAN kesilmelidir.
Hipokalsemi:
Diferansiye tiroid kanserli hastalarda sorafenib kullan�l�yorken, kan kalsiyum d�zeyinin yak�ndan izlenmesi �nerilmektedir. Klinik �al��malarda hipokalsemi; diferansiye tiroid kanserli hastalarda (�zellikle hipoparatiroidizm �yk�s� olanlarda), renal h�creli veya
hepatosel�ler kanserli hastalar ile kar��la�t�r�ld���nda daha s�k ve daha �iddetli g�r�lm��t�r (bkz. B�l�m 4.8).
DTK'de tiroid stim�le edici hormon (TSH) supresyonu:
Sorafenib eksojen tiroid supresyonunu olumsuz etkiler. DTK �al��mas�nda, hastalar�n
%99'unun ba�lang��taki tiroid stim�le edici hormon (TSH) d�zeyi 0,5 mU/L'nin alt�ndad�r. 0,5 mU/L'nin �zerine ��kan TSH d�zeyleri, sorafenib ile tedavi edilen hastalar�n %41'inde, plasebo ile tedavi edilen hastalar�n %16's�nda g�zlemlenmi�tir. Sorafenib kullan�rken TSH supresyonunda bozulma g�r�len hastalarda, medyan maksimum TSH 1,6 mU/L olup, hastalar�n %25'inde TSH d�zeyi 4,4 mU/L'nin �zerinde kaydedilmi�tir.
Diferansiye tiroid kanserli hastalarda sorafenib kullan�l�yorken, TSH d�zeyinin yak�ndan izlenmesi ve gerekirse tiroid replasman tedavisinde ayarlama yap�lmas� �nerilmektedir.
�la� etkile�imleri:
UGT1A yolu: Sorafenib, esas olarak UGT1A1 yolu veya UGT1A9 yolu arac�l���yla metabolize veya elimine edilen bile�ikler (�rn. irinotekan) ile birlikte uygulan�rken dikkatli olunmas� �nerilmektedir (bkz. B�l�m 4.5).
Dosetaksel: Dosetaksel (75 ya da 100 mg/m) ile birlikte sorafenib (g�nde iki kez 200 ya da 400 mg) uygulamas� (dosetaksel uygulamas� s�ras�nda sorafenibe 3 g�n ara verme �eklinde), dosetaksel EAA de�erinde %36-80 art�� ile sonu�lanm��t�r. Sorafenib dosetaksel ile birlikte uygulan�rken dikkatli olunmas� �nerilmektedir (bkz. B�l�m 4.5).
Neomisin: E� zamanl� neomisin uygulamas� ve ba��rsak floras�n� bozan di�er antibiyotiklerin uygulanmas� sorafenibin biyoyararlan�m�nda azalmaya neden olabilir (bkz. Di�er t�bbi �r�nler ile etkile�imler). Antibiyotiklerle bir tedavi k�r�ne ba�lanmadan �nce, plazma sorafenib konsantrasyonlar�n�n azalma riski de�erlendirilmelidir.
Akci�er karsinomunda platin-bazl� kemoterapi s�ras�nda sorafenib kullan�lmas� y�ksek mortaliteye neden olur. K���k h�creli d��� akci�er kanseri tan�l� hastalar�n de�erlendirildi�i iki randomize �al��mada, skuam�z karsinomlu hastalar�n olu�turdu�u alt grupta paklitaksel/karboplatine ek olarak sorafenib tedavisi alanlarda genel sa�kal�m i�in HR 1,81 (%95 GA 1,19; 2,74) olarak belirlenirken, gemsitabin/sisplatine ek tedavi i�in 1,22 (%95 GA 0,82; 1,8) olmu�tur. Tekli �l�m nedenlerinden hi�biri bask�n olmam��; ancak, platin bazl� kemoterapilere ek olarak sorafenib uygulanan hastalarda solunum yetmezli�i, kanamalar ve enfeksiy�z advers olaylar bak�m�ndan daha y�ksek bir insidans kaydedilmi�tir.
Hastal��a �zel uyar�lar
Diferansiye tiroid kanseri (DTK):
Hekimlere, tedaviyi ba�latmadan �nce maksimum lezyon boyutu (bkz. B�l�m 5.1), hastal�kla ilgili semptomlar (bkz. B�l�m 5.1) ve progresyon h�z�n� dikkate alarak hastadaki prognozu dikkatli bir �ekilde de�erlendirmesi �nerilmektedir.
��pheli advers ila� reaksiyonlar�n�n y�netimi sorafenib tedavisinin ge�ici olarak kesilmesini veya dozunun azalt�lmas�n� gerektirebilir. �al��ma 5'de (bkz. B�l�m 5.1), sorafenib tedavisinin hen�z 1. siklusunda g�n�ll�lerin %37'sinde doza ara verilmesi ve %35'inde dozun azalt�lmas� gerekmi�tir.
Doz azalt�m�, advers reaksiyonlar�n hafifletilmesinde yaln�zca k�smen ba�ar�l� olmu�tur. Bu
nedenle, anti-t�m�r aktivitesi ve tolerabilite de dikkate al�narak tekrarlanan fayda/risk de�erlendirmeleri �nerilmektedir.
DTK'de hemoraji:
Potansiyel kanama riski nedeniyle, DTK hastalar�na sorafenib uygulanmadan �nce trakeal, bron�iyal ve �zefageal infiltrasyon i�in lokal tedavi uygulanmal�d�r.
DTK'de hipokalsemi:
DTK hastalar�nda sorafenib kullan�l�rken kan kalsiyum d�zeyinin yak�ndan izlenmesi �nerilir.
Klinik �al��malarda, �zellikle hipoparatiroidizm �yk�s� olanlar olmak �zere DTK hastalar�nda hipokalsemi, renal h�creli veya hepatosell�ler karsinomu olan hastalara g�re daha s�k ve daha �iddetli olmu�tur. Derece 3 ve 4 hipokalsemi, sorafenib ile tedavi edilen DTK hastalar�n�n %6,8'i ve %3,4'�nde geli�mi�tir (bkz. B�l�m 4.8). QT uzamas� veya Torsade de pointes gibi komplikasyonlar�n �nlenmesi i�in (bkz. B�l�m 4.4.) �iddetli hipokalsemi d�zeltilmelidir.
DTK'de TSH supresyonu:
�al��ma 5'de (bkz. B�l�m 5.1), sorafenib ile tedavi edilen hastalar�n TSH d�zeylerinde 0,5 mU/L'nin �zerinde art��lar g�zlemlenmi�tir. DTK hastalar�nda sorafenib kullan�l�rken TSH d�zeyinin yak�ndan izlenmesi �nerilir.
Renal h�creli karsinom:
MSKCC (Memorial Sloan Kettering Cancer Center) prognostik grubuna g�re Y�ksek Riskli Hastalar, renal h�creli karsinoma ili�kin faz III klinik �al��maya dahil edilmemi� (bkz. B�l�m 5.1), dolay�s�yla bu hastalarda fayda-risk de�erlendirmesi yap�lmam��t�r.
4.5. Di�er t�bbi �r�nler ile etkile�imler ve di�er etkile�im �ekilleri
CYP3A4 ind�kleyicileri:
Tek doz sorafenib uygulanmadan �nce 5 g�n s�reyle rifampisin uygulanmas� sorafenib EAA de�erinde ortalama %37'lik bir azalmayla sonu�lanm��t�r. CYP3A4 aktivitesinin ve/veya glukuronidasyonunun di�er ind�kleyicileri de (�rn. St. John's wort Hypericum perforatum bitkisi, fenitoin, karbamazepin, fenobarbital ve deksametazon), sorafenib metabolizmas�n� art�rabilir ve b�ylelikle sorafenib konsantrasyonlar�n� azaltabilirler.
CYP3A4 inhibit�rleri:
G��l� bir CYP3A4 inhibit�r� olan ketokonazol, sa�l�kl� erkek g�n�ll�lere 7 g�n s�reyle g�nde bir kez uyguland���nda, tek doz 50 mg sorafenibin ortalama EAA de�erini de�i�tirmemi�tir. Bu nedenle sorafenib ve CYP3A4 inhibit�rleri aras�nda klinik farmakokinetik etkile�im olas�l��� pek bulunmamaktad�r.
CYP2B6, CYP2C8 ve CYP2C9 substratlar�:
Sorafenib, in vitro ko�ullarda CYP2B6, CYP2C8 ve CYP2C9'u benzer potensle inhibe etmi�tir. Bununla birlikte, klinik farmakokinetik �al��malar�nda g�nde iki kez sorafenib 400 mg'�n bir CYP2B6 substrat� olan siklofosfamid veya bir CYP2C8 substrat� olan paklitaksel ile birlikte uygulanmas� klinik olarak anlaml� inhibisyonla sonu�lanmam��t�r. Bu veriler, �nerilen doz olan g�nde iki kez 400 mg sorafenibin CYP2B6 veya CYP2C8'in in vivo inhibit�r� olmayabilece�ini d���nd�rmektedir.
Sorafenib in vitro ko�ullarda CYP2B6 ve CYP2C8'i s�ras�yla 6 ve 1-2 µM Ki de�erleri ile inhibe etmektedir.
Ayr� bir klinik �al��mada, sorafenibin paklitaksel ile e� zamanl� uygulamas�, CYP2C8 taraf�ndan olu�turulan etkin paklitaksel metaboliti 6-OH paklitaksel maruziyetinde azalma yerine art��a neden olmu�tur. Bu veriler sorafenibin in vivo olarak CYP2C8 inhibit�r� olmayabilece�ini ileri s�rmektedir. Farkl� bir klinik �al��mada sorafenibin siklofosfamid ile e�zamanl� kullan�m� siklofosfamid maruziyetinde k���k bir azalma ile sonu�lanm��t�r. Ancak CYP2B6 taraf�ndan olu�turulan siklofosfamidin etkin metaboliti 4-OH siklofosfamidin sistemik maruziyetinde bir azalma meydana gelmemi�tir. Bu veriler sorafenibin, CYP2B6'n�n in vivo inhibit�r� olmayabilece�ini i�aret etmektedir.
�nsan karaci�er mikrozomlar� ile �al��malar sorafenibin 7-8 µM Ki de�eri ile CYP2C9'un kompetitif inbibit�r� oldu�unu g�stermi�tir. Ayr�ca sorafenib ve bir CYP2C9 substrat� olan varfarin ile e� zamanl� tedavi, ortalama PT-INR'de plaseboya g�re de�i�iklik olu�turmam��t�r. Dolay�s�yla, CYP2C9'un sorafenib taraf�ndan klinik olarak ilgili in vivo inhibisyonu riskinin de d���k olmas� beklenebilir. Ancak, varfarin veya fenprokumon kullanan hastalar�n INR'leri d�zenli olarak kontrol edilmelidir (bkz. B�l�m 4.4).
CYP3A4, CYP2D6 ve CYP2C19 substratlar�:
S�ras�yla CYP3A4, CYP2D6 ve CYP2C19 sitokromlar�n�n substratlar� olan sorafenib ve midazolam, dekstrometorfan veya omeprazol�n e� zamanl� uygulamas� bu ajanlara maruz kalmay� de�i�tirmemi�tir. Bu durum, sorafenibin bu sitokrom P450 izoenzimlerinin inhibit�r� veya ind�kleyicisi olmad���n� g�stermektedir. Bu nedenle, sorafenibin bu enzimlerin substratlar� ile klinik farmakokinetik etkile�imi �ng�r�lmemektedir.
UGT1A1 ve UGT1A9 substratlar�:
�n vitro ko�ullarda, sorafenib UGT1A1 ve UGT1A9 arac�l���yla glukuronidasyonu inhibe etmi�tir. Bu bulgunun klinik ilgisi bilinmemektedir (bkz. B�l�m 4.4). Sorafenibin irinotekan ile birlikte klinik olarak kullan�m� sonucu SN-38 olan aktif metaboliti UGT1A1 arac�l���yla tekrardan metabolize edilir ve sonucunda SN-38'in EAA'�nda %67-120 art�� g�zlenir.
CYP enzim ind�ksiyonuna ili�kin in vitro ko�ullarda yap�lm�� �al��malar:
CYP1A2 ve CYP3A4'�n aktiviteleri, k�lt�r� yap�lan insan hepatositlerinin sorafenib ile tedavisinden sonra de�i�memi�tir, bu da sorafenibin CYP1A2 ve CYP3A4'�n ind�kleyicisi olma olas�l���n�n d���k oldu�unu g�stermektedir.
P-gp substratlar�:
�n vitro, sorafenibin transport protein p-glikoproteini (P-gp) inhibe etti�i g�sterilmi�tir. Sorafenib ile e� zamanl� tedavi sonucunda digoksin gibi P-gp substratlar�n�n plazma konsantrasyonlar�nda art�� olas�l��� d��lanamaz.
Di�er antineoplastik ajanlar ile kombinasyon:
Sorafenib klinik �al��malarda, gemsitabin, sisplatin, oksaliplatin, paklitaksel, karboplatin, kapesitabin, doksorubisin, irinotekan, dosetaksel ve siklofosfamid gibi �e�itli di�er anti- neoplastik ajanlar ile birlikte, bu ajanlar�n s�kl�kla kullan�lan dozaj rejimleriyle kullan�lm��t�r.
Sorafenib gemsitabin, sisplatin, karboplatin, oksaliplatin veya siklofosfamid farmakokineti�i �zerinde klinik olarak anlaml� bir etki g�stermemi�tir.
Paklitaksel/Karboplatin
Paklitaksel/karboplatin uygulamas� s�ras�nda sorafenib dozunda 3 g�nl�k bir ara verilerek, paklitaksel (225 mg/m) ve karboplatinin (EAA=6) sorafenible (g�nde iki kez ≤400 mg) birlikte uygulanmas�, paklitaksel farmakokinetik de�erlerinde anlaml� herhangi bir etki olu�turmam��t�r.
Paklitaksel (225 mg/m, her 3 haftada bir kez) ve karboplatinin (EAA=6) sorafenible (g�nde iki kez 400 mg, sorafenib dozunda bir ara olmaks�z�n) birlikte uygulanmas�, sorafenib maruziyetinde %47'lik, paklitaksel maruziyetinde %29'luk ve 6-OH paklitaksel maruziyetinde ise %50'lik bir art�� olu�turmu�tur. Karboplatin farmakokinetik de�erleri etkilenmemi�tir.
Bu veriler, paklitaksel ve karboplatinin sorafenib dozunda 3 g�nl�k bir ara dikkate al�narak sorafenible birlikte uygulanmas� durumunda doz ayarlamas�na gerek olmad���n� ortaya koymaktad�r. Ara olmaks�z�n verilen e� zamanl� sorafenibi takiben sorafenib ve paklitaksel maruziyetlerinde g�zlenen art���n klinik anlam� bilinmemektedir.
Kapesitabin
E� zamanl� kapesitabin (750-1050 mg/m-g�nde iki kez, her 21 g�nl�k s�re i�inde 1-14. g�nlerde) ve sorafenib (g�nde iki kez 200 ya da 400 mg, s�rekli-kesintisiz uygulama) uygulamas�, sorafenib maruziyetinde anlaml� bir de�i�iklik olu�turmam��; ancak kapesitabin maruziyetinde %15-50'lik, 5-FU maruziyetinde ise %0-52'lik bir art��la sonu�lanm��t�r. Sorafenible e�zamanl� uyguland���nda kapesitabin ve 5-FU maruziyetlerinde g�zlenen bu hafif ila orta d�zeyli art��lar�n klinik anlam� bilinmemektedir.
Doksorubisin/�rinotekan
Sorafenib ile e� zamanl� uygulama, doksorubisinin EAA de�erinde %21'lik bir art��la sonu�lanm��t�r. Aktif metaboliti SN-38'in daha sonra UGT1A1 yoluyla metabolize oldu�u irinotekan ile birlikte uyguland���nda, SN-38'in EAA de�erinde %67-120, irinotekan�n EAA de�erinde ise %26-42 art�� vard�r. Bu bulgular�n klinik �nemi bilinmemektedir (bkz. B�l�m 4.4).
Dosetaksel
Dosetaksel (21 g�nde bir uygulanan 75 ya da 100 mg/m) ile sorafenibin (21 g�nl�k siklusun
2. g�n�nden 19. g�n�ne kadar, g�nde iki kez 200 ya da 400 mg; dosetaksel uygulamas� s�ras�nda sorafenibe 3 g�n ara verme �eklinde) birlikte uygulanmas�, dosetakselin EAA de�erinde %36-80 ve dosetakselin Cd�zeyinde %16-32 art�� ile sonu�lanm��t�r. Sorafenib ile dosetakselin birlikte uygulanmas� s�ras�nda dikkatli olunmas� �nerilmektedir (bkz. B�l�m 4.4).
Antibiyotikler ile kombinasyon
Neomisin
Gastrointestinal floray� eradike etmek i�in kullan�lan sistemik olmayan bir antimikrobiyal ajan olan neomisinin e� zamanl� uygulamas�, sorafenibin enterohepatik geri d�n���m�n� engeller (bkz. B�l�m 5.2) ve bunun sonucunda sorafenib maruziyetinde d����e neden olur. 5 g�n s�reli neomisin rejimiyle tedavi g�ren sa�l�kl� g�n�ll�lerde ortalama sorafenib maruziyeti
%54 azalm��t�r. Bu bulgular�n klinik anlam� bilinmemektedir. Di�er antibiyotiklerin etkileri ara�t�r�lmam��t�r ancak b�y�k olas�l�kla glukuronidaz aktivitesini azaltma yetenekleriyle ili�kilendirileceklerdir.
Proton pompas� inhibit�rleri ile kombinasyon
Omeprazol
Omeprazol ile birlikte kullan�m�n sorafenibin farmakokinetik de�erleri �zerinde herhangi bir etkisi yoktur. Sorafenib i�in bir doz ayarlamas� yap�lmas� gerekmez.
�zel pop�lasyonlara ili�kin ek bilgiler
Veri bulunmamaktad�r.
Pediyatrik pop�lasyon:
Veri bulunmamaktad�r.
4.6. Gebelik ve laktasyon
Gebelik kategorisi: D
�ocuk do�urma potansiyeli bulunan kad�nlar/Do�um kontrol� (Kontrasepsiyon) Hayvanlar �zerinde yap�lan ara�t�rmalar sorafenibin teratojenik ve embriyotoksik oldu�unu g�stermi�tir. �ocuk do�urma potansiyeli olan kad�nlar tedavi s�resince ve tedavinin ard�ndan en az 2 haftaya kadar, etkili do�um kontrol� uygulamak zorundad�rlar (bkz. B�l�m 4.4, B�l�m 5.3).
Kad�nlar tedavi alt�nda iken gebe kalmaktan ka��nmal�d�r. �ocuk do�urma potansiyeline sahip kad�nlara fet�s �zerindeki a��r malformasyon (teratojenisite), b�y�me gerili�i ve fetal �l�m (embriyotoksisite) gibi potansiyel tehlikeler bildirilmelidir.
Gebelik d�nemi
Sorafenibin gebelik ve/veya fet�s/yenido�an �zerinde zararl� farmakolojik etkileri bulunmaktad�r. Sorafenib kullanan gebe kad�nlar �zerinde y�r�t�lm�� yeterli ve iyi kontroll� �al��malar bulunmamaktad�r. Hayvanlarda y�r�t�len �al��malarda malformasyonlar� i�eren reprod�ktif toksisite g�sterilmi�tir (bkz. B�l�m 4.4). S��anlarda sorafenib ve metabolitlerinin plasentaya ge�tikleri bulunmu�tur ve sorafenibin fetusta anjiyojenezi inhibe etmesi beklenmektedir.
SOFEXAN gebelik s�ras�nda kullan�lmamal�d�r. Doktorlar bu ilac�n kullan�m�n�, sadece potansiyel yararlar� fet�s �zerindeki potansiyel riskleri hakl� ��kar�yorsa g�ndeme getirebilirler (bkz. B�l�m 4.4, B�l�m 5.3).
Laktasyon d�nemi
Sorafenibin insan s�t� ile at�l�p at�lmad��� bilinmemektedir.
Hayvanlar �zerinde yap�lan �al��malar sorafenib ve/veya metabolitlerinin s�t ile at�ld���n� g�stermektedir. Sorafenibin fet�s�n b�y�mesine ve geli�mesine zararl� etkileri olabilece�inden, sorafenib tedavisi s�ras�nda emzirme b�rak�lmal�d�r.
�reme yetene�i/Fertilite
Hayvan �al��malar�n�n sonu�lar�, sorafenibin erkek ve kad�nlarda fertiliteyi bozabilece�ine i�aret etmektedir (bkz. B�l�m 5.3).
4.7. Ara� ve makine kullan�m� �zerindeki etkiler
Sorafenib ile ara� ya da makine kullanma yetileri �zerindeki etkilerine y�nelik �al��ma yap�lmam��t�r. Sorafenibin ara� ya da makine kullanma yetilerini etkiledi�ine ili�kin bir veri bulunmamaktad�r.
4.8. �stenmeyen etkiler
En �nemli ciddi advers reaksiyonlar, miyokard enfarkt�s�/iskemisi, gastrointestinal perforasyon, ila�la ind�klenen hepatit, hemoraji ve hipertansiyon/hipertansif kriz olmu�tur.
En yayg�n advers reaksiyonlar diyare, yorgunluk, alopesi, enfeksiyon, el-ayak deri reaksiyonu (MedDRA'da palmar-plantar eritrodizestezi sendromuna kar��l�k gelir), d�k�nt�, kilo kayb�, i�tah kayb�, mide bulant�s�, gastrointestinal ve abdominal a�r�, hipertansiyon ve hemoraji olmu�tur.
Birden fazla klinik �al��ma s�ras�nda meydana gelen ya da pazarlama sonras� kullan�m s�resince saptanan t�m advers reaksiyonlar, a�a��da sistem-organ s�n�f� (MedDRA) ve s�kl�k derecesine g�re listelenmektedir. S�kl�k dereceleri �u �ekilde tan�mlanmaktad�r; �ok yayg�n (≥1/10); yayg�n (≥1/100 ila <1/10); yayg�n olmayan (≥1/1.000 ila <1/100); seyrek (≥1/10.000 ila <1/1.000); �ok seyrek (<1/10.000); bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor).
Her s�kl�k grubu i�inde, istenmeyen etkiler azalan �iddet derecesine g�re s�ralanm��t�r.
Enfeksiyonlar ve enfestasyonlar
�ok yayg�n: Enfeksiyon Yayg�n: Folik�lit
Kan ve lenf sistemi hastal�klar�
�ok yayg�n: Lenfopeni
Yayg�n: L�kopeni, n�tropeni, anemi, trombositopeni
Ba����kl�k sistemi hastal�klar�
Yayg�n olmayan: Anafilaktik reaksiyon, a��r� duyarl�l�k reaksiyonlar� (deri reaksiyonlar� ve �rtiker dahil)
Seyrek: Anjiyo�dem
Endokrin hastal�klar�
Yayg�n: Hipotiroidizm
Yayg�n olmayan: Hipertiroidizm
Metabolizma ve beslenme hastal�klar�
�ok yayg�n: Anoreksi, hipofosfatemi
Yayg�n: Hipokalsemi, hipokalemi, hiponatremi, hipoglisemi Yayg�n olmayan: Dehidrasyon
Bilinmiyor: T�m�r lizis sendromu
Psikiyatrik hastal�klar
Yayg�n: Depresyon
Sinir sistemi hastal�klar�
Yayg�n: Periferik duyusal n�ropati, disguzi
Yayg�n olmayan: Geri d�n���ml� posterior l�koensefalopati* Bilinmiyor: Ensefalopati
Kulak ve i�kulak hastal�klar�
Yayg�n: Tinnitus
Kardiyak hastal�klar
Yayg�n: Konjestif kalp yetmezli�i*, miyokard iskemisi ve/veya enfarkt�s�* Seyrek: QT uzamas�
Vask�ler hastal�klar
�ok yayg�n: Hemoraji (gastrointestinal*, solunum yolu* ve serebral hemoraji* dahil), hipertansiyon
Yayg�n: S�cak basmas� (flushing) Yayg�n olmayan: Hipertansif kriz*
Bilinmiyor: Anevrizmalar ve arter diseksiyonlar�
Solunum, g���s bozukluklar� ve mediyastinal hastal�klar
Yayg�n: Rinore, disfoni
Yayg�n olmayan: �nterstisyel akci�er hastal��� benzeri olaylar* (pn�monit, radyasyon pn�moniti, akut solunum g��l���, interstisyel pn�moni, pulmonit ve akci�er inflamasyonu bildirimleri de dahil olmak �zere)
Gastrointestinal hastal�klar
�ok yayg�n: Diyare, bulant�, kusma, konstipasyon
Yayg�n: Stomatit (a��z kurulu�u ve glossodini dahil), dispepsi, disfaji, gastro�zofajiyal refl� hastal���
Yayg�n olmayan: Pankreatit, gastrit, gastrointestinal perforasyonlar*
Hepatobiliyer hastal�klar
Yayg�n olmayan: Bilir�bin art��� ve sar�l�k, kolesistit, kolanjit Seyrek: �laca ba�l� hepatit*
Deri ve derialt� dokusu hastal�klar�
�ok yayg�n: Deri kurulu�u, d�k�nt�, alopesi, el-ayak deri reaksiyonu**, pruritus, eritem Yayg�n: Deride keratoakantomalar/skuam�z h�creli kanser, eksfolyatif dermatit, akne, deri deskuamasyonu, hiperkeratoz
Yayg�n olmayan: Egzama, eritema multiforme
Seyrek: Radyasyon dermatiti, l�kositoklastik vask�lit, Stevens-Johnson sendromu, toksik epidermal nekroliz*
Kas-iskelet bozukluklar�, ba� dokusu ve kemik hastal�klar�
�ok yayg�n: Artralji
Yayg�n: Miyalji, kas spazmlar� Seyrek: Rabdomiyoliz
B�brek ve idrar yolu hastal�klar�
Yayg�n: B�brek yetmezli�i, protein�ri
Seyrek: Nefrotik sendrom
�reme sistemi ve meme hastal�klar�
Yayg�n: Erektil disfonksiyon Yayg�n olmayan: Jinekomasti
Genel bozukluklar ve uygulama b�lgesine ili�kin hastal�klar
�ok yayg�n: Yorgunluk, a�r� (a��z, abdominal, kemik, t�m�r a�r�s� ve ba� a�r�s� dahil), ate� Yayg�n: Asteni, grip benzeri hastal�k, mukozal inflamasyon
Ara�t�rmalar
�ok yayg�n: Kilo kayb�, amilaz art���, lipaz art��� Yayg�n: Transaminazlarda ge�ici art��
Yayg�n olmayan: Kan alkalen fosfataz�nda ge�ici art��, anormal INR (Uluslararas� Normalizasyon Oran�), protrombin d�zeyinde anormallik
* Bu advers reaksiyonlar hayat� tehdit edici ya da �l�mc�l sonu� verebilir. Bu gibi olaylar ya yayg�n olmayan ya da yayg�n olmayandan daha az s�kl�ktad�r.
**MedDRA'da palmar plantar eritrodisestezi sendromuna uygun d��mektedir.
Baz� advers ila� reaksiyonlar� ile ilgili ilave bilgi
Konjestif Kalp Yetmezli�i - Y�r�t�len klinik �al��malarda, konjestif kalp yetmezli�i sorafenib tedavisi g�ren hastalar�n (n=2.276) %1,9'unda advers olay olarak bildirilmi�tir. Renal h�creli karsinoma (RHK) �al��mas� 11213'te (�al��ma 1) konjestif kalp yetmezli�iyle uyumlu advers olaylar sorafenib tedavisi g�ren hastalar�n %1,7'sinde ve plasebo grubunun %0,7'sinde bildirilmi�tir. Hepatosel�ler karsinoma (HSK) �al��mas� 100554'de (�al��ma 3), sorafenib tedavisi g�renlerin %0,99'unda, plasebo kullananlar�n ise %1,1'inde bu olaylar bildirilmi�tir.
�leri k���k h�cre d��� akci�er karsinomu (KHDAK) olan hastalar�n ilk basamak tedavisi olarak sorafenibin ikili platinyum-bazl� kemoterapilerle (karboplatin/paklitaksel ve ayr� ayr� gemsitabin/sisplatin) kombine kullan�m�na kar��l�k sadece ilgili ikili platinyum-bazl� kemoterapinin g�venlilik ve etkilili�inin kar��la�t�r�ld��� iki randomize plasebo kontroll� �al��ma, genel sa�kal�m�n iyile�mesi olan birincil sonlan�m noktas�na ula�amam��t�r. G�venlilik olaylar� genellikle daha �nce bildirilen olaylarla uyumludur. Ancak, iki �al��mada da tek ba��na ikili platinyum-bazl� kemoterapi (karboplatin/paklitaksel ve ayr� ayr� gemsitabin/sisplatin) tedavisiyle kar��la�t�r�ld���nda, sorafenib ve ikili platinyum-bazl� kemoterapi tedavisi uygulanan skuam�z h�creli akci�er kanseri olan bir hasta alt grubunda daha y�ksek mortalite izlenmi�tir (HR 1,22, %95 GA 0,82-1,8). Bu bulgulara ili�kin kesin bir gerek�e tan�mlanmam��t�r.
G�venlilik ayn� zamanda bir faz II �al��ma havuzunda da de�erlendirilmi�tir. Bu toplu veriler, sorafenib ile tedavi edilen 638 hastaya aittir ve bunlar�n aras�nda 202 hastada renal h�creli karsinoma, 137 hastada hepatosel�ler karsinoma ve 299 hastada ba�ka kanser t�rleri bulunmaktad�r. Bu toplu veriler i�inde sorafenib tedavisindeki hastalarda en s�k bildirilen ilaca ba�l� advers olaylar; d�k�nt� (%38), diyare (%37), el-ayak deri reaksiyonu (%35) ve yorgunluk (%33) olmu�tur. Sorafenib tedavisindeki hastalarda ortak toksisite kriterleri (CTC) (v 2.0) Derece 3 ve 4 ilaca ba�l� advers olaylar, s�ras�yla %37 ve %3'd�r.
Klinik �al��malarda, renal h�creli karsinoma veya hepatosel�ler karsinoma �al��malar�nda yer alan hastalar ile kar��la�t�r�ld���nda diferansiye tiroid kanserli hastalarda el-ayak deri
reaksiyonu, diyare, alopesi, kilo kayb�, hipertansiyon, hipokalsemi ve deride keratoakantomalar/skuam�z h�creli kanser gibi belli advers ila� reaksiyonlar� b�y�k �l��de daha y�ksek s�kl�kta ortaya ��km��t�r.
RHK hastalar�nda laboratuvar test anormallikleri (�al��ma 11213) (�al��ma 1):
Lipaz ve amilaz d�zeylerinde art�� s�k olarak bildirilmi�tir. �al��ma 11213'te, Advers olaylar i�in ortak terminoloji kriterleri (CTCAE) derece 3 ya da 4 lipaz art��lar�, sorafenib grubundaki hastalar�n %12'sinde bildirilirken, plasebo grubundaki hastalar�n %7'sinde bildirilmi�tir. CTCAE derece 3 ya da 4 amilaz art��lar�, sorafenib grubundaki hastalar�n %1'inde bildirilirken, plasebo grubundaki hastalar�n %3'�nde bildirilmi�tir. �al��ma 1'de klinik pankreatit, sorafenib tedavisindeki 451 hastan�n 2'sinde (CTCAE derece 4) ve plasebo grubundaki 451 hastan�n 1'inde (CTCAE derece 2) bildirilmi�tir.
Hipofosfatemi s�k rastlanan laboratuvar anomalisidir, sorafenib tedavisindeki hastalar�n
%45'inde, plasebo hastalar�n�n %11'inde g�zlenmi�tir. CTCAE derece 3 hipofosfatemi (1-2 mg/dL), sorafenib ile tedavi edilen hastalar�n %13'�nde ve plasebo grubundaki hastalar�n
%3'�nde ortaya ��km��t�r. CTCAE derece 4 hipofosfatemi (<1 mg/dL) olgusu sorafenib ve plasebo kollar�nda bildirilmemi�tir. Sorafenib ile ili�kili hipofosfateminin etiyolojisi bilinmemektedir.
CTCAE derece 3 ya da 4 olaylar, lenfopeni i�in sorafenib tedavisindeki hastalar�n %13'�nde ve plasebo hastalar�n�n %7'sinde, n�tropeni i�in sorafenib tedavisindeki hastalar�n %5'inde ve plasebo hastalar�n�n %2'sinde, anemi i�in sorafenib tedavisindeki hastalar�n %2'sinde ve plasebo hastalar�n�n %4'�nde ve trombositopeni i�in sorafenib tedavisindeki hastalar�n %1 ve plasebo hastalar�n�n %0'�nda bildirilmi�tir.
Sorafenib ile tedavi edilen hastalar�n %12'sinde hipokalsemi bildirilirken plasebo alan hastalarda bu oran %7,5 olmu�tur. Bildirilen �o�u hipokalsemi d���k derecededir (CTCAE Derece 1 ve 2). CTCAE derece 3 hipokalsemi (6-7 mg/dL) sorafenib ile tedavi edilen hastalar�n %1,1'inde, plasebo grubundaki hastalar�n ise %0,2'sinde g�r�lm��; CTCAE derece
4 hipokalsemi de (<6 mg/dL) sorafenib ile tedavi edilen hastalar�n %1,1'inde, plasebo grubundaki hastalar�n ise %0,5'inde g�r�lm��t�r. Sorafenib ile ili�kili hipokalseminin etiyolojisi bilinmemektedir.
Sorafenib ile tedavi edilen hastalar�n %5,4'�nde hipokalemi bildirilirken, plasebo alan hastalarda bu oran %0,7 olmu�tur. Bildirilen �o�u hipokalemi d���k derecededir (CTCAE derece 1). CTCAE derece 3 hipokalemi, sorafenib ile tedavi edilen hastalar�n %1,1'inde, plasebo grubundaki hastalar�n ise %0,2'sinde g�r�lm��t�r. Derece 4 hipokalemi bildirilmemi�tir.
HSK hastalar�nda laboratuvar anormallikleri (�al��ma 100554):
Lipaz art��� sorafenib ile tedavi edilen hastalar�n %40'�nda g�zlenirken, plasebo grubundaki hastalar�n %37'sinde g�zlenmi�tir. CTCAE derece 3 ya da 4 lipaz art��lar�, her iki grupta da hastalar�n %9'unda ortaya ��km��t�r. Amilaz art��lar�, sorafenib ile tedavi edilen hastalar�n
%34'�nde g�zlenirken, plasebo grubundaki hastalar�n %29'unda g�zlenmi�tir. CTCAE derece 3 ya da 4 amilaz art��lar�, her iki grupta da hastalar�n %2'sinde bildirilmi�tir. Lipaz ve amilaz art��lar�n�n �o�u ge�icidir ve olgular�n b�y�k �o�unlu�unda sorafenib tedavisine ara verilmemi�tir. Klinik pankreatit sorafenib tedavisindeki 297 hastan�n 1'inde bildirilmi�tir (CTCAE derece 2).
Hipofosfatemi s�k rastlanan bir laboratuvar bulgusudur, sorafenib ile tedavi edilen hastalar�n
%35'inde, plasebo hastalar�n�n %11'inde g�zlenmi�tir. CTCAE derece 3 hipofosfatemi (1-2 mg/dL), sorafenib ile tedavi edilen hastalar�n %11'inde ve plasebo grubundaki hastalar�n
%2'sinde ortaya ��km��t�r; plasebo grubunda bildirilen 1 CTCAE derece 4 hipofosfatemi (<1 mg/dL) olgusu bulunmaktad�r. Sorafenib ile ili�kili hipofosfateminin etiyolojisi bilinmemektedir.
Karaci�er fonksiyon testlerindeki art��lar, �al��man�n 2 kolu aras�nda kar��la�t�r�labilir niteliktedir. Aspartat aminotransferaz (AST) art��lar� sorafenib ile tedavi edilen hastalar�n
%94'� ve plasebo hastalar�n�n %91'inde g�zlenmi�tir. CTCAE derece 3 ya da 4 AST art��lar�, sorafenib ile tedavi edilen hastalar�n %16's�nda ve plasebo grubundaki hastalar�n %17'sinde bildirilmi�tir. ALT art��lar� sorafenib ile tedavi edilen hastalar�n %69'unda ve plasebo hastalar�n�n %68'inde g�zlenmi�tir. CTCAE derece 3 ya da 4 alanin aminotransferaz (ALT) art��lar�, sorafenib ile tedavi edilen hastalar�n %3'�nde ve plasebo tedavisindeki hastalar�n
%8'inde bildirilmi�tir. Bilir�bin art��lar�, sorafenib ile tedavi edilen hastalar�n %47'sinde ve plasebo hastalar�n�n %45'inde g�zlenmi�tir. CTCAE derece 3 ya da 4 bilir�bin art��lar� sorafenib ile tedavi edilen hastalar�n %10'unda ve plasebo tedavisindeki hastalar�n %11'inde bildirilmi�tir. Hipoalb�minemi, sorafenib ile tedavi edilen hastalar�n %59'unda ve plasebo hastalar�n�n %47'sinde g�zlenmi�tir; her iki grupta da CTCAE derece 3 ya da 4 hipoalb�minemi g�zlenmemi�tir.
Alkalen fosfataz art��lar�, sorafenib ile tedavi edilen hastalar�n %82,2'sinde ve plasebo hastalar�n�n %82,5'inde g�zlenmi�tir. CTCAE derece 3 alkalen fosfataz art��lar�, sorafenib ile tedavi edilen hastalar�n %6,2'sinde ve plasebo tedavisindeki hastalar�n %8,2'sinde bildirilmi�tir; her iki grupta da hi� CTCAE derece 4 alkalen fosfataz art��� g�zlenmemi�tir.
INR art��lar�, sorafenib ile tedavi edilen hastalar�n %42'sinde ve plasebo hastalar�n�n
%34'�nde g�zlenmi�tir. CTCAE derece 3 INR art��lar� sorafenib ile tedavi edilen hastalar�n
%4'�nde ve plasebo hastalar�n�n %2'sinde bildirilmi�tir; her iki grupta da CTCAE derece 4 INR y�kselmesi bulunmamaktad�r.
Lenfopeni, sorafenib ile tedavi edilen hastalar�n %47'sinde ve plasebo hastalar�n�n %42'sinde g�zlenmi�tir. CTCAE derece 3 ya da 4 lenfopeni, her bir gruptaki hastalar�n %6's�nda bildirilmi�tir. N�tropeni sorafenib ile tedavi edilen hastalar�n %11'inde ve plasebo hastalar�n�n %14'�nde g�zlenmi�tir. CTCAE derece 3 ya da 4 n�tropeni, her bir gruptaki hastalar�n %1'inde bildirilmi�tir.
Anemi, sorafenib ile tedavi edilen hastalar�n %59'unda ve plasebo hastalar�n�n %64'�nde g�zlenmi�tir. CTCAE derece 3 ya da 4 anemi, her bir gruptaki hastalar�n %3'�nde bildirilmi�tir.
Trombositopeni, sorafenib ile tedavi edilen hastalar�n %46's�nda ve plasebo hastalar�n�n
%41'inde g�zlenmi�tir. CTCAE derece 3 ya da 4 trombositopeni sorafenib ile tedavi edilen hastalar�n %4'�nde ve plasebo hastalar�n�n %1'den daha az�nda bildirilmi�tir.
Sorafenib ile tedavi edilen hastalar�n %26,5'inde hipokalsemi bildirilirken plasebo alan hastalarda bu oran %14,8 olmu�tur. Bildirilen �o�u hipokalsemi d���k derecededir (CTCAE derece 1 ve 2). CTCAE derece 3 hipokalsemi (6-7 mg/dL) sorafenib ile tedavi edilen hastalar�n %1,8'inde, plasebo grubundaki hastalar�n ise %1,1'inde g�r�lm��; CTCAE derece
4 hipokalsemi de (<6 mg/dL) sorafenib ile tedavi edilen hastalar�n %0,4'�nde, plasebo
grubundaki hastalar�n ise %0'�nda g�r�lm��t�r. Sorafenib ile ili�kili hipokalseminin etiyolojisi bilinmemektedir.
Sorafenib ile tedavi edilen hastalar�n %9,5'inde hipokalemi bildirilirken, plasebo alan hastalarda bu oran %5,9 olmu�tur. Bildirilen �o�u hipokalemi d���k derecededir (CTCAE derece 1). CTCAE derece 3 hipokalemi, sorafenib ile tedavi edilen hastalar�n %0,4'�nde, plasebo grubundaki hastalar�n ise %0,7'sinde g�r�lm��t�r. Derece 4 hipokalemi bildirilmemi�tir.
Tiroid karsinoma hastalar�nda laboratuvar test anormallikleri (�al��ma 5):
Sorafenib ile tedavi edilen hastalar�n %35,7'sinde hipokalsemi bildirilirken, plasebo grubundaki hastalarda bu oran %11 olmu�tur. Bildirilen �o�u hipokalsemi d���k derecededir. CTCAE derece 3 hipokalsemi sorafenib ile tedavi edilen hastalar�n %6,8'inde, plasebo grubundaki hastalar�n %1,9'unda; CTCAE derece 4 hipokalsemi ise sorafenib ile tedavi edilen hastalar�n %3,4'�nde, plasebo grubundaki hastalar�n %1'inde g�r�lm��t�r.
�al��mada g�zlemlenen di�er klinik olarak anlaml� laboratuvar anormallikleri, Tablo 4' de g�sterilmektedir.
Tablo 4: Diferansiye Tiroid Kanserli Hastalarda Bildirilen Tedaviye Ba�l� Laboratuvar Testi Anormallikleri_�ift K�r D�nemi (�al��ma 5)
Laboratuvar parametresi, (% ara�t�r�lan numuneler) | Sorafenib, n=207 | Plasebo, n=209 | ||||
T�m Dereceler* | Derece 3* | Derece 4* | T�m Dereceler * | Derece 3* | Derece 4* | |
Kan ve lenf sistemi hastal�klar� | ||||||
Anemi | 30,9 | 0,5 | 0 | 23,4 | 0,5 | 0 |
Trombositopeni | 18,4 | 0 | 0 | 9,6 | 0 | 0 |
N�tropeni | 19,8 | 0,5 | 0,5 | 12 | 0 | 0 |
Lenfopeni | 42 | 9,7 | 0,5 | 25,8 | 5,3 | 0 |
Metabolizma ve beslenme hastal�klar� | ||||||
Hipokalemi | 17,9 | 1,9 | 0 | 2,4 | 0 | 0 |
Hipofosfatemi** | 19,3 | 12,6 | 0 | 2,4 | 1,4 | 0 |
Hepatobiliyer hastal�klar | ||||||
Bilirubin art��� | 8,7 | 0 | 0 | 4,8 | 0 | 0 |
ALT art��� | 58,9 | 3,4 | 1 | 24,4 | 0 | 0 |
AST art��� | 53,6 | 1 | 1 | 14,8 | 0 | 0 |
Ara�t�rmalar | ||||||
Amilaz art��� | 12,6 | 2,4 | 1,4 | 6,2 | 0 | 1 |
Lipaz art��� | 11,1 | 2,4 | 0 | 2,9 | 0,5 | 0 |
* Advers Olaylar i�in Ortak Terminoloji Kriteri (CTCAE), Versiyon 3.0
** Sorafenib ile ili�kili hipofosfateminin etiyolojisi bilinmemektedir.
��pheli advers reaksiyonlar�n raporlanmas�
Ruhsatland�rma sonras� ��pheli ila� advers reaksiyonlar�n�n raporlanmas� b�y�k �nem ta��maktad�r. Raporlama yap�lmas�, ilac�n yarar/risk dengesinin s�rekli olarak izlenmesine olanak sa�lar. Sa�l�k mesle�i mensuplar�n�n herhangi bir ��pheli advers reaksiyonu T�rkiye Farmakovijilans Merkezi (T�FAM)'ne bildirmeleri gerekmektedir (www.titck.gov.tr; e-
posta: tufam@titck.gov.tr; tel: 0 800 314 00 08; faks: 0 312 218 35 99).
4.9. Doz a��m� ve tedavisi
SOFEXAN doz a��m� i�in spesifik bir tedavi bulunmamaktad�r.
Klinik olarak incelenen en y�ksek sorafenib dozu g�nde iki kez 800 mg'd�r. Bu dozda g�zlenen advers reaksiyonlar ba�l�ca diyare ve dermatolojik olaylar olmu�tur.
Bir doz a��m� ku�kusu durumunda, SOFEXAN uygulamas� durdurulmal� ve destekleyici tedavi ba�lat�lmal�d�r.
5. FARMAKOLOJ�K �ZELL�KLER
5.1. Farmakodinamik �zellikler
Farmakoterap�tik grup: Antineoplastik ila�lar, protein kinaz inhibit�r� ATC kodu: L01EX02
Sorafenib, in vitro ve in vivo anti-proliferatif ve anti-anjiyojenik �zellikler sergileyen bir multikinaz inhibit�r�d�r.
Etki mekanizmas� ve farmakodinamik etkiler
Sorafenib, in vitro ko�ullarda t�m�r h�cresi proliferasyonunu azaltan bir multikinaz inhibit�r�d�r. Sorafenib, atimik farelerde t�m�r anjiyogenezinde azalma ile birlikte, insan t�m�r� ksenograftlar�n�n geni� bir spektrumunda t�m�r b�y�mesini inhibe etmektedir. Sorafenib t�m�r h�cresinde (CRAF, BRAF, V600E BRAF, c-KIT ve FLT-3) ve t�m�r vask�lat�r�nde (CRAF, VEGFR-2, VEGFR-3 ve PDGFR-ß) bulunan hedeflerin aktivitesini inhibe etmektedir. Serin/treonin kinazlar RAF kinazlar� iken c-KIT, FLT-3, VEGFR-2, VEGFR-3 ve PDGFR-ß resept�r tirozin kinazlar�d�r.
Klinik etkililik ve g�venlilik
Sorafenibin klinik etkilili�i ve g�venlili�i hepatosel�ler karsinomal� (HSK), metastatik renal h�creli karsinomal� (RHK) ve diferansiye tiroid karsinomal� (DTK) hastalarda �al���lm��t�r.
Hepatosel�ler karsinoma
�al��ma 3 (�al��ma 100554), 602 hepatosell�ler karsinom hastas�yla yap�lan uluslararas�, �ok merkezli, randomize, �ift k�rl�, plasebo kontroll� bir Faz III �al��mad�r. Sorafenib ve plasebo grubunda ECOG skoru a��s�ndan demografik ve ba�lang��taki hastal�k �zellikleri benzerdir (skor 0: %54'e kar�� %54; skor 1: %38'e kar�� %39; skor 2: %8'e kar�� %7), TNM evresi (evre I: <%1'e kar�� <%1; evre II: %10,4'e kar�� %8,3; evre III: %37,8'e kar�� %43,6; evre IV: %50,8'e kar�� %46,9) ve BCLC evresi (evre B: %18,1'e kar�� %16,8; evre C: %81,6'ya kar�� %83,2; evre D: <%1'e kar�� %0).
�al��ma, planlanan ara OS analizi �nceden belirlenen etkililik s�n�r�n� ge�tikten sonra durdurulmu�tur. Bu OS analizi, sorafenibin OS a��s�ndan plaseboya g�re istatistiksel olarak anlaml� �l��de avantajl� oldu�unu g�stermi�tir (HR: 0,69, p = 0,00058, bkz. Tablo 5).
Bu �al��mada Child Pugh B karaci�er yetmezli�i olan hastalara ili�kin veriler s�n�rl�d�r ve Child Pugh C olan yaln�zca bir hasta �al��maya dahil edilmi�tir.
Tablo 5: Hepatosell�ler Karsinomda �al��ma 3 (�al��ma 100554) Etkililik Sonu�lar�
Etkililik Parametresi | Sorafenib (n=299) | Plasebo (n=303) | P de�eri | HR (%95 GA) |
Genel Sa�kal�m (OS) [medyan, hafta (%95 GA)] | 46,3
(40,9; 57,9) | 34,4
(29,4; 39,4) | 0,00058* | 0,69
(0,55; 0,87) |
�lerlemeye Kadar Ge�en Zaman (TTP) [medyan, hafta (%95 GA)]** | 24
(18; 30) | 12,3
(11,7; 17,1) |
0,000007 | 0,58
(0,45; 0,74) |
GA=G�ven aral���, HR=Tehlike oran� (plaseboya g�re Sorafenib)
*p de�eri, �nceden belirlenen O'Brien Fleming durma s�n�r� olan 0,0077'in alt�nda oldu�undan istatistiksel olarak anlaml�
**Ba��ms�z radyolojik inceleme
�kinci Faz III, uluslararas�, �ok merkezli, randomize, �ift k�rl�, plasebo kontroll� �al��mada (�al��ma 4, 11849) ilerlemi� hepatosell�ler karsinomu olan 226 hastada sorafenibin klinik faydas� de�erlendirilmi�tir. �in, Kore ve Tayvan'da y�r�t�len bu �al��ma, sorafenibin olumlu fayda-risk profili a��s�ndan �al��ma 3'�n bulgular�n� do�rulam��t�r (HR (OS): 0,68, p = 0,01414).
�al��ma 3 ve 4'te �nceden belirlenen katmanland�rma fakt�rlerinde (ECOG skoru, makroskopik vask�ler invazyon ve/veya ekstrahepatik t�m�r yay�lmas� varl��� veya yoklu�u) tehlike oran� tutarl� bir �ekilde plaseboya g�re sorafenib lehine olmu�tur. Ara�t�rma ama�l� alt grup analizleri, ba�lang��ta uzak metastazlar� olan hastalarda tedavi etkisinin daha az belirgin oldu�unu g�stermi�tir.
Renal h�creli karsinoma
Sorafenibin ilerlemi� renal h�creli karsinoma (RHK) tedavisindeki g�venlili�i ve etkilili�i 2 klinik �al��mada incelenmi�tir:
�al��ma 1 (�al��ma 11213), 903 hastayla yap�lan �ok merkezli, randomize, �ift k�rl�, plasebo kontroll� bir Faz III �al��mad�r. Yaln�zca berrak h�creli renal karsinomu ve d���k ve orta MSKCC (Memorial Sloan Kettering Kanser Merkezi) riski olan hastalar �al��maya dahil edilmi�tir. Primer sonlan�m noktalar� genel sa�kal�m ve progresyonsuz sa�kal�m (PFS) olmu�tur.
Hastalar�n yakla��k yar�s�nda ECOG performans skoru 0'd�r ve hastalar�n yar�s� d���k risk MSKCC prognostik grubundad�r.
PFS, RECIST kriterleri kullan�larak k�rle�tirilmi� ba��ms�z radyolojik inceleme ile de�erlendirilmi�tir. PFS analizi 769 hastada 342 olayla y�r�t�lm��t�r. Medyan PFS, sorafenibe randomize edilen hastalarda 167 g�n olarak bulunurken, plasebo hastalar�nda 84 g�n olmu�tur (HR=0,44; %95 GA: 0,35-0,55; p<0,000001). Ya�, MSKCC prognostik grubu, ECOG PS ve �nceki tedavi, tedavinin etki boyutunu etkilememi�tir.
Genel sa�kal�ma y�nelik ara analiz (ikinci ara analiz) 903 hastada 367 �l�mle yap�lm��t�r. Bu analiz i�in nominal alfa de�eri 0,0094't�r. Medyan sa�kal�m, sorafenibe randomize edilen hastalarda 19,3 ay olarak bulunurken, plasebo hastalar�nda 15,9 ay olmu�tur (HR= 0,77; %95 GA: 0,63, 0,95; p = 0,015). Bu analiz s�ras�nda yakla��k 200 hasta plasebo grubundan
sorafenib grubuna ge�mi�tir.
�al��ma 2, RHK'y� da i�eren metastatik maligniteleri olan hastalarla yap�lan bir Faz II, kesme �al��mas�d�r. Sorafenib tedavisinde hastal��� stabil olan hastalar plaseboya randomize edilmi� veya sorafenib tedavisine devam etmi�tir. RHK's� olan hastalarda progresyonsuz sa�kal�m (PFS), sorafenib grubunda (163 g�n) plasebo grubuna (41 g�n) g�re anlaml� �l��de daha uzun olmu�tur (p= 0,0001, HR= 0,29).
Diferansiye tiroid karsinoma
�al��ma 5 (�al��ma 14295), radyoaktif iyoda diren�li lokal olarak ilerlemi� veya metastatik diferansiye tiroid kanserli 417 hasta �zerinde y�r�t�len, uluslararas�, �ok-merkezli, randomize, �ift-k�r, plasebo-kontroll� bir Faz III ara�t�rmad�r.
K�rlenmi� bir ba��ms�z radyolojik inceleme ile RECIST kriterleri kullan�larak de�erlendirilen PFS, �al��man�n birincil sonlan�m noktas�d�r. Sekonder sonlan�m noktalar� aras�nda genel sa�kal�m (OS), t�m�r yan�t oran� ve yan�t s�resi yer alm��t�r. Progresyon sonras�nda, hastalar�n a��k etiketli sorafenib kullanmalar�na izin verilmi�tir.
Kay�ttan �nce 14 ay i�inde hastalarda progresyon g�r�ld�yse ve Radyoaktif iyota (RA�) diren�li DTK varsa hastalar �al��maya dahil edilmi�tir. RA�'ye diren�li DTK RA� taramas�nda iyot tutulumu olmayan bir lezyonu olan veya k�m�latif RA�>600 mCi alan veya kay�ttan �nceki 16 ay i�inde bir RA� tedavisi ya da 16 ayl�k s�re i�inde yap�lm�� iki RA� tedavisinden sonra progresyon g�steren olarak tan�mlanm��t�r.
Ba�lang�� d�nemi demografik �zellikleri ve hasta �zellikleri her iki tedavi grubu aras�nda iyi bir dengelenme g�stermi�tir. Hastalar�n %86's�nda akci�erlerde, %51'inde lenf nodunda ve
%27'sinde kemikte metastaz vard�r. Hastalar�n hemen hemen tamam�na tiroidektomi (%99,5) uygulanm�� ve ortanca yakla��k 400 mCi k�m�latif radyoaktif aktivite alm��t�r. Hastalar�n �o�unda papiller karsinomay� (%56,8) takiben folik�ler (%25,4) ve daha az diferansiye karsinoma (%9,6) vard�r.
T�m analiz k�mesinde, 207 hasta g�nde iki kez 400 mg sorafenibe ve 210 hasta plaseboya randomize edilmi�tir. PFS, k�rlemeli ba��ms�z radyolojik inceleme yoluyla, RECIST kriterleri kullan�larak de�erlendirilmi�tir.
Ortanca PFS s�resi, sorafenib grubunda 329 g�n (10,8 ay) ve plasebo grubunda 175 g�nd�r (5,8 ay). PFS i�in ba��l risk (hastal�k progresyonu veya �l�m), sorafenib ile tedavi edilen hastalarda, plasebo ile tedavi edilen bireyler ile kar��la�t�r�ld���nda tehlike oran�yla yakla��k
%41 azalm��t�r (Tehlike Oran� (TO): 0,587; %95 GA: 0,454; 0,758; tek y�nl� p<0,0001)
(Tablo 6, �ekil 2).
Sorafenibin PFS �zerindeki etkisi co�rafik b�lge, 60 ya� �zeri veya alt� ya�, cinsiyet, histolojik alt tip, t�m�r y�k� ve kemik metastaz�n�n varl��� veya yoklu�u dahil t�m alt k�melerde tutarl�d�r.
Genel sa�kal�mda (OS) tedavi gruplar� aras�nda istatistiksel bir fark bulunmamaktad�r (TO: 0,802; %95 GA: 0,539; 1,194, tek y�nl� p de�eri 0,138, Tablo 6). Ortanca OS'ye her iki kolda da eri�ilmemi�tir. Plasebo koluna randomize edilen 150 (%71,4) ve sorafenib koluna randomize edilen 55 (%26,6) hasta a��k etiketli olarak sorafenib ald�lar.
RECIST'e g�re tam yan�t (CR) g�zlenmemi�tir. Ba��ms�z radyolojik de�erlendirmeye g�re
genel yan�t oran� (CR + parsiyel yan�t (PR)) sorafenib grubunda (24 hasta, %11,6), plasebo grubuna (1 hasta, %0,5) k�yasla daha y�ksektir (tek y�nl� p<0,0001). PR veren, sorafenib ile tedavi edilen hastalarda ortalama yan�t s�resi 309 g�nd�r (%95 GA: 226, 505 g�n).
Tablo 6: �al��ma 5: Diferansiye Tiroid Karsinomada Etkililik Sonu�lar�
Etkinlik Parametresi | Sorafenib (n=207) Olay (�l�m) ile deneklerin (%) oran� | Plasebo (n=210) Olay (�l�m) ile deneklerin (%) oran� | Sorafenib (n=207) [ortanca, g�n (%95 GA)]* | Plasebo (n=210) [ortanca, g�n (%95 GA)] | p-de�eri | TO (%95 GA) |
Progresyonsuz Sa�kal�m (PFS) (12.08.2012) | 113 (%54,6) | 137 (%65,2) | 329 (278; 393) | 175 (160; 238) | <0,0001 | 0,587 (0,454-0,758) |
Genel Sa�kal�m (OS) (31.05.2013) | 66 (%31,9) | 72 (%34,3) | Eri�ilmemi�tir | 1.110 (26-1.110) | 0,2359 | 0,884 (0,663; 1,236) |
GA=G�ven aral���, TO=Tehlike oran� (plaseboya kar�� sorafenib)
* Ba��ms�z radyolojik inceleme
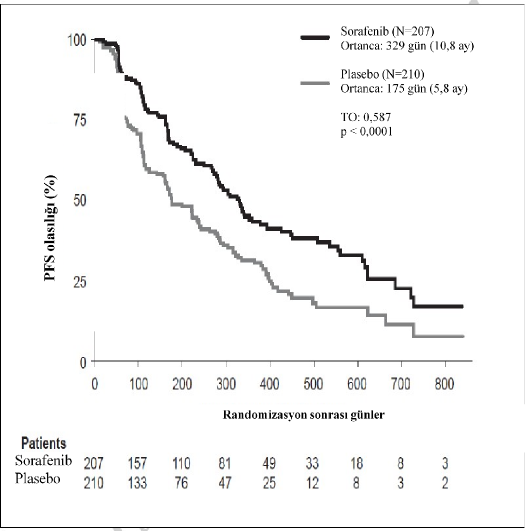
Riskli hastalar
�ekil 1: �al��ma 5: Tiroid Karsinomada Progresyonsuz Sa�kal�m�n (PFS) Kaplan-Meier e�risi (t�m analiz k�mesi)
QT aral���n�n uzamas�
Bir klinik farmakoloji �al��mas�nda, 31 hastada ba�lang��ta (tedavi �ncesi) ve tedavi sonras� QT/QTc �l��mleri kaydedilmi�tir. 28 g�nl�k tek bir tedavi k�r�nden sonra, maksimum sorafenib konsantrasyonunun izlendi�i zaman noktas�nda, plasebo tedavisinin ba�lang�� de�erleriyle kar��la�t�r�ld���nda QTcB 4±19 msn, QTcF ise 9±18 msn uzam��t�r. Tedavi sonras� EKG izlemi s�ras�nda hi�bir hastada QTcB veya QTcF>500 msn izlenmemi�tir (bkz. B�l�m 4.4).
5.2. Farmakokinetik �zellikler
Genel �zelliklerEmilim:
Sorafenib tablet uygulamas�ndan sonra, oral ��zeltiye k�yasla ortalama ba��l biyoyararlan�m
%38-49'dur. Oral uygulamay� izleyerek, sorafenib doruk plazma d�zeylerine yakla��k 3 saatte ula��r. Orta dereceli ya� i�eren bir yemek ile verildi�inde, biyoyararlan�m� a� kar�na oldu�u gibidir. Ya�dan zengin bir yemek ile verildi�inde, sorafenib biyoyararlan�m� a� kar�na uygulamaya k�yasla %30 azalmaktad�r. Yedi g�n s�reyle �oklu sorafenib dozlar�n�n uygulanmas�, tek doz uygulamaya k�yasla 2,5 ile 7 katl�k bir birikim ile sonu�lan�r. Kararl� durum plazma sorafenib konsantrasyonlar� 7 g�n i�inde elde edilir ve ortalama konsantrasyonlar�n tepe-vadi oran� 2'den d���kt�r. G�nde iki kez 400 mg sorafenib uygulamas�n�n kararl� durum farmakokineti�i, tiroid karsinoma, RHK ve HSK hastalar�nda de�erlendirilmi�tir. T�m t�m�r tipleri i�in maruz kalmadaki de�i�kenlik y�ksek olmas�na ra�men, en y�ksek ortalama maruziyet tiroid kanserli hastalarda g�r�lm��t�r. Tiroid kanserli hastalarda EAA'daki art���n klinik anlam� bilinmemektedir.
Da��l�m:
Sorafenibin insan plazma proteinlerine in vitro ba�lanmas� %99,5 d�zeyindedir.
Biyotransformasyon:
Sorafenib esas olarak karaci�erde metabolize olur, CYP3A4'�n arac�l�k etti�i oksidatif metabolizmaya girerken, ayn� zamanda UGT1A9 arac�l���yla glukuronidasyona u�rar. Sorafenib konjugatlar� gastrointestinal kanalda bakteriyel glukuronidaz aktivitesi taraf�ndan par�alanabilirler, bu da konjuge olmayan ilac�n tekrar emilimini sa�lar. E� zamanl� neomisin uygulamas� bu s�reci engelleyerek sorafenibin ortalama biyoyararlan�m�n� %54 oran�nda d���r�r.
Sorafenib kararl� durumda, plazmada dola�an metabolitlerin yakla��k %70-85'ini olu�turur. Sorafenibin sekiz metaboliti tan�mlanm��t�r, bunlardan be�i plazmada saptanm��t�r. Sorafenibin plazmada dola�an esas metaboliti, piridin N-oksit, sorafenibe benzer bir in vitro potens g�sterir. Bu metabolit kararl� durumda dola��mdaki metabolitlerin yakla��k %9-16's�n� olu�turur.
Eliminasyon:
��zelti �eklinde bir sorafenib form�lasyonu oral yoldan 100 mg dozda uyguland�ktan sonra dozun %96's� 14 g�n i�inde at�lm��t�r, bu miktar�n %77'si fe�es ile, %19'u idrarda glukuronize metabolitler �eklinde at�lm��t�r. Dozun %51'ini olu�turan de�i�memi� haldeki sorafenib, fe�este bulunmakta, ama idrarda bulunmamaktad�r. Bu da, de�i�memi� haldeki ilac�n eliminasyonuna safra ile at�l�m�n katk�da bulundu�unu g�stermektedir.
Sorafenibin eliminasyon yar�-�mr� 25-48 saat civar�ndad�r.
Do�rusall�k/Do�rusal olmayan durum:
Sorafenib do�rusal farmakokinetik �zellik g�sterir.
Hastalardaki karakteristik �zellikler
Irk:
Beyaz �rk ile Asya �rk�na mensup g�n�ll�lerde farmakokinetik �zellikler bak�m�ndan klinik olarak anlaml� farkl�l�klar bulunmamaktad�r.
Cinsiyet:
Demografik verilerin analizleri, cinsiyete g�re farmakokinetik a��s�ndan fark olmad���n� g�stermektedir.
Karaci�er yetmezli�i:
Sorafenib esas olarak karaci�er taraf�ndan elimine edilmektedir.
Hafif (Child-Pugh A) ya da orta derecede (Child-Pugh B) karaci�er yetmezli�i olan hepatosel�ler karsinomu hastalarda sistemik maruz kalma d�zeyleri, karaci�er bozuklu�u olmayan hastalarda g�zlenen aral�k i�indedir. Child-Pugh A ve Child-Pugh B olup hepatosel�ler karsinomu olmayan hastalarda sorafenibin farmakokineti�i, sa�l�kl� g�n�ll�lerin farmakokineti�i ile benzerdir.
Sorafenib farmakokineti�i �iddetli karaci�er yetmezli�i (Child-Pugh C) olan hastalarda incelenmemi�tir (bkz. �zel kullan�m uyar�lar� ve �nlemleri ve Pozoloji ve uygulama �ekli). Ba�l�ca karaci�er arac�l���yla at�ld���ndan bu hasta pop�lasyonunda maruziyet artabilir.
B�brek yetmezli�i:
Bir klinik farmakoloji �al��mas�nda sorafenibin farmakokineti�i, b�brek fonksiyonlar� normal olan olgulara ve hafif (CrCl 50-80 mL/dk), orta dereceli (CrCl 30 ile <50 mL/dk) ya da diyaliz gerektirmeyen �iddetli b�brek yetmezli�i (CrCl <30 mL/dk) olan olgulara tek doz 400 mg uygulamas�ndan sonra de�erlendirilmi�tir. Sistemik sorafenibe maruz kalma ve renal fonksiyon aras�nda ili�ki g�zlenmemi�tir. Hafif, orta dereceli ya da diyaliz gerektirmeyen �iddetli b�brek yetmezli�i nedeniyle doz ayarlamas� gerekli de�ildir (bkz. Pozoloji ve uygulama �ekli). Diyaliz gereken hastalardaki durum incelenmemi�tir.
Ya�l�lar (65 ya� �zeri):
Demografik verilerin analizleri, ya�a g�re doz ayarlamas� yap�lmas�n�n gerekli olmad���n� g�stermektedir.
Pediyatrik hastalar:
Pediyatrik hastalara ili�kin farmakokinetik veri bulunmamaktad�r.
5.3. Klinik �ncesi g�venlilik verileri
Karsinojenez, Mutajenez, Fertilite bozuklu�u
Sorafenibin klinik �ncesi g�venlilik profili fareler, s��anlar, k�pekler ve tav�anlarda de�erlendirilmi�tir.
Tekrarl� doz toksisitesi �al��malar�nda, �ng�r�len klinik maruziyetin alt�ndaki maruziyetlerde (EAA kar��la�t�rmalar� do�rultusunda) �e�itli organlarda de�i�iklikler (dejenerasyon ve rejenerasyon) g�r�lm��t�r.
Gen� ve b�y�mekte olan k�peklere tekrarl� doz uygulamas�ndan sonra, kemik ve di�ler �zerinde etkiler g�zlenmi�tir. Bu de�i�iklikler, g�nl�k 600 mg/m v�cut y�zey alan� sorafenib dozunda (v�cut y�zey alan� temelinde �nerilen klinik doz olan 500 mg/m'nin 1,2 kat�na e�de�er) femoral b�y�me pla��nda d�zensiz kal�nla�malar, 200 mg/m/g�n d�zeyinde de�i�en b�y�me pla��na kom�u kemik ili�inde hiposel�larite ve 600 mg/m/g�n d�zeyinde dentin bile�iminde de�i�ikliklerden olu�maktayd�. Eri�kin k�peklerde benzeri etkiler ind�klenmemi�tir.
Bir in vitro memeli h�cresi �al��mas�nda (�in hamsteri overleri), metabolik aktivasyon varl���nda klastojenisite (kromozomal aberrasyonlar) i�in pozitif genotoksik etkiler elde edilmi�tir. �retim prosesi s�ras�nda olu�an ve ayn� zamanda bitmi� ila� hammaddesinde de bulunan (<%0,15) bir ara madde, bir in vitro bakteriyel h�cre incelemesinde (Ames testi) mutajenez a��s�ndan pozitiftir. Sorafenib, Ames testinde (%0,34 d�zeyinde ara madde) ve bir in vivo ko�ullarda fare mikronukleus incelemesinde genotoksik bulunmam��t�r.
Sorafenib ile karsinogenesite �al��malar� y�r�t�lmemi�tir.
Sorafenib ile hayvanlarda fertilite �zerindeki etkiyi de�erlendirme ama�l� spesifik �al��malar yap�lmam��t�r. Ancak erkek ve di�i fertilitesi �zerinde bir advers etki beklenebilir, ��nk� hayvanlarda y�r�t�len tekrarl� doz �al��malar�, erkek ve di�i �reme organlar�nda de�i�imler oldu�unu g�stermi�tir. Tipik de�i�imler, s��anlar�n testis, epididim, prostat ve seminal vezik�llerinde dejenerasyon ve retardasyon bulgular�ndan olu�maktad�r ve bu etkiler g�nl�k 150 mg/m v�cut y�zey alan� sorafenib dozunda a��k bir �ekilde belirmi�tir (v�cut y�zey alan� temelinde �nerilen 500 mg/m'lik klinik dozun yakla��k ��te biri). Di�i s��anlarda korpus luteumda santral nekroz ve overlerde folik�ler geli�im duraklamas� g�r�lm��t�r ve g�zlenen en d���k etki d�zeyi 30 mg/m/g�n olmu�tur. K�peklerde 600 mg/m/g�n dozunda testislerde t�b�ler dejenerasyon ve 1200 mg/m/g�n dozunda oligospermi g�r�lm��t�r.
Sorafenibin s��anlar ve tav�anlara uyguland���nda embriyotoksik ve teratojen oldu�u g�sterilmi�tir. G�zlenen etkiler maternal ve fetal v�cut a��rl���nda azalma, fetal rezorpsiyon say�s�nda art�� ve eksternal ve viseral malformasyon say�s�nda art��tan olu�maktad�r. S��anlarda oral 6 mg/m/g�n dozunda ve tav�anlarda 36 mg/m/g�n dozunda advers fetal sonlan�mlar g�zlenmi�tir (bkz. B�l�m 4.4, B�l�m 4.6).
�evresel Risk de�erlendirme �al��malar�, sorafenib tosilat�n �evre a��s�ndan persistan, biyoak�m�latif ve toksik olma potansiyeli oldu�unu g�stermi�tir.
6. FARMAS�T�K �ZELL�KLER
6.1. Yard�mc� maddelerin listesi
Mikrokristalin sel�loz PH101 Mikrokristalin sel�loz PH102 Kroskarmelloz sodyum
Hidroksi propil metilsel�loz E5 LV premium Sodyum lauril s�lfat
Magnezyum stearat Polietilen glikol 6000
Opadry complete film coating system 03B240041 pink (hipromelloz, titanyum dioksit, makrogol/PEG, k�rm�z� demiroksit)
6.2. Ge�imsizlikler
Bilinen bir ge�imsizli�i bulunmamaktad�r.
6.3. Raf �mr�
24 ay
6.4. Saklamaya y�nelik �zel tedbirler
SOFEXAN 25°C'nin alt�ndaki oda s�cakl���nda ambalaj�nda saklanmal�d�r.
6.5. Ambalaj�n niteli�i ve i�eri�i
�effaf PVC-PE-PVDC-Al�minyum folyo blister ambalajlarda 112 film kapl� tablet
6.6. Be�eri t�bbi �r�nden arta kalan maddelerin imhas� ve di�er �zel �nlemler
SOFEXAN isimli t�bbi �r�n �evre i�in potansiyel risk olu�turabilir.
Kullan�lmam�� olan �r�nler ya da at�k materyaller, “T�bbi At�klar�n Kontrol� Y�netmeli�i” ve “Ambalaj At�klar�n�n Kontrol� Y�netmeli�i”ne uygun olarak imha edilmelidir.
 Y�ksek Tansiyon
Hipertansiyon s�rekli anormal derecede y�ksek olan kan bas�nc�d�r. Tansiyon
atardamarlar�n�zdaki kan�n bas�nc�d�r.
Y�ksek Tansiyon
Hipertansiyon s�rekli anormal derecede y�ksek olan kan bas�nc�d�r. Tansiyon
atardamarlar�n�zdaki kan�n bas�nc�d�r. |
 S�rt A�r�s�
S�rt a�r�s� birden bire ortaya
��k�p �iddetli (akut) olabilir veya zamanla geli�ip daha uzun
s�reli sorunlara (kronik) neden olabilir.
S�rt A�r�s�
S�rt a�r�s� birden bire ortaya
��k�p �iddetli (akut) olabilir veya zamanla geli�ip daha uzun
s�reli sorunlara (kronik) neden olabilir. |
�LA� E�DE�ERLER�
| E�de�er �la� Ad� | Barkodu | �la� Fiyat� |
|---|---|---|
| PROKINIB | 8699702099027 | 42,033.80TL |
| SOFEXAN | 8699540033658 | 42,033.80TL |
| Di�er E�de�er �la�lar |
 |
Ast�m Ast�ml� ki�ilerin akci�erlerindeki hava borular� (bron�lar) hassast�r. Bu ki�iler belirli tetikleyici fakt�rlere maruz kald�klar�nda, hava borular� nefes almalar�n� g��le�tirecek �ekilde daral�r. |
 |
En Yayg�n Alerji T�rleri Ba����kl�k sistemi, polen, ar� zehiri veya evcil hayvan gibi yabanc� bir maddeye veya �o�u insanda reaksiyona neden olmayan bir yiyece�e tepki g�sterdi�inde alerjiler meydana gelir. |
 |
A��z Kanseri A��z kanserinin en yayg�n t�rleri, dudak, dil, di�etidir. Nadiren yanak i�i veya damak b�lgelerini de i�ine al�r. |
�LA� GENEL B�LG�LER�
Nobel �la� Sanayii ve Tic. Anomim �irketi
| Sat�� Fiyat� | 42033.8 TL [ 3 Aug 2026 ] |
| �nceki Sat�� Fiyat� | 42033.8 TL [ 27 Jul 2026 ] |
| Original / Jenerik | Original �la� |
| Re�ete Durumu | Normal Re�eteli bir ila�d�r. |
| Barkodu | 8699540033658 |
| Etkin Madde | Sorafenib |
| ATC Kodu | L01EX02 |
| Birim Miktar | 200 |
| Birim Cinsi | MG |
| Ambalaj Miktar� | 112 |
| Antineoplastik ve �mm�nomod�lat�r Ajanlar |
| Yerli ve Be�eri bir ila�d�r. |