SIMKELMA 5G oral s�spansiyon haz�rlamak i�in toz-11 sa�e K�sa �r�n Bilgisi
{ Sodyum Zirkonyum Siklosilikat }
1. BE�ER� TIBB� �R�N�N ADI
SIMKELMA 5 g oral s�spansiyon haz�rlamak i�in toz
2. KAL�TAT�F VE KANT�TAT�F B�LE��M
Etkin madde
Her bir sa�e 5 g sodyum zirkonyum siklosilikat i�erir.
Yard�mc� maddeler
SIMKELMA, yard�mc� madde i�ermemektedir.
3. FARMAS�T�K FORMU
Oral s�spansiyon haz�rlamak i�in toz. Beyaz ila gri aras� toz.
4. KL�N�K �ZELL�KLER
4.1. Terap�tik endikasyonlar
SIMKELMA, eri�kin hastalarda hiperkalemi tedavisinde endikedir (Bkz. B�l�m 4.4 ve B�l�m 5.1).
4.2. Pozoloji ve uygulama �ekli
Eri�kinlerD�zeltme faz�
SIMKELMA'n�n �nerilen ba�lang�� dozu, su i�inde s�spansiyon halinde oral yolla g�nde �� kez uygulanan 10 g'd�r. Normokalemiye ula��ld���nda idame rejimine ge�ilmesi gerekmektedir (a�a��daki a��klamaya bak�n�z).
Normokalemiye genel olarak 24 ila 48 saat i�inde ula��l�r. Hastalar, 48 saatlik tedaviden sonra halen hiperkalemik ise, ayn� rejime 24 saat daha devam edilebilir. E�er 72 saatlik tedaviden sonra normokalemiye ula��lam�yorsa ba�ka tedavi yakla��mlar�n�n g�z �n�nde bulundurulmas� gerekmektedir.
�dame faz�
S�rekli idame tedavisinde, hiperkalemi n�ks�n� �nlemek i�in minimum etkili doz olu�turulmal�d�r. Normal bir potasyum seviyesini korumak i�in, g�nde bir kez 5 g'l�k doz �nerilir, gerekirse g�nde birkez10g'akadardozartt�r�labilir veya g�n a��r� 5 g'a kadar doz
Tedavi s�ras�nda serum potasyum d�zeylerinin d�zenli olarak izlenmesi gerekmektedir. �zlem s�kl���; di�er ila�lar, kronik b�brek hastal���n�n progresyonu ve diyetle al�nan potasyum miktar� gibi �e�itli fakt�rlere ba�l� olacakt�r.
E�er �iddetli hipokalemi meydana gelirse, SIMKELMA kesilmeli ve hasta yeniden de�erlendirilmelidir.
Kronik hemodiyaliz uygulanan hastalar
Diyaliz g�rmekte olan hastalara SIMKELMA yaln�zca diyaliz uygulanmayan g�nlerde verilmelidir. �nerilen ba�lang�� dozu g�nde bir defa 5 g'dir. Normokalemi (4,0-5,0 mmol/L) sa�lamak amac�yla doz, uzun diyaliz aras�ndan (LIDI) sonra �l��len diyaliz �ncesi serum potasyum de�erine ba�l� olarak haftal�k olarak yukar� veya a�a�� titre edilebilir. Doz, bir haftal�k aral�klarla ve 5 g'lik basamaklarla diyaliz uygulanmayan g�nlerde g�nde bir defa 15 g'ye kadar ayarlanabilir. Doz ayarland��� s�rada serum potasyum seviyelerinin her hafta kontrol edilmesi �nerilir; normokalemi sa�land���nda potasyum d�zenli �ekilde (�rne�in her ay veya diyetsel potasyum veya serum potasyum d�zeyini etkileyen ila�lar� i�eren klinik de�erlendirmeye ba�l� olarak daha s�k) takip edilmelidir.
Unutulan doz
Hasta bir dozu almay� unutursa, kendisine bir sonraki dozu normal zaman�nda almas� s�ylenmelidir.
Uygulama �ekli:
SIMKELMA oral kullan�m i�indir. S�spansiyon a� veya tok karn�na al�nabilir.
S�spansiyonun haz�rlanmas�na ili�kin talimatlar i�in bkz. b�l�m 6.6.
�zel pop�lasyonlara ili�kin ek bilgiler:
B�brek yetmezli�i:
B�brek yetmezli�i olan hastalar i�in normal dozlarda herhangi bir de�i�iklik gerekmemektedir.
Karaci�er yetmezli�i:
Karaci�er yetmezli�i olan hastalar i�in normal dozlarda herhangi bir de�i�iklik gerekmemektedir.
Pediyatrik pop�lasyon:
SIMKELMA'n�n g�venlili�i ve etkilili�i 18 ya��n alt�ndaki ergenlerde ve �ocuklarda belirlenmemi�tir. Veri bulunmamaktad�r.
Geriyatrik pop�lasyon:
Geriyatrik hastalar i�in doz ayarlamas� gerekmez.
4.3. Kontrendikasyonlar
SIMKELMA, sodyum zirkonyum siklosilikata kar�� a��r� duyarl�l��� olan hastalarda kontrendikedir.
4.4. �zel kullan�m uyar�lar� ve �nlemleri
Serum potasyum d�zeyleri
Serum potasyum konsantrasyonunu etkileyen t�bbi �r�nlerde (�rn. renin-anjiyotensin- aldosteron sistemi (RAAS) inhibit�rleri veya di�retikler) de�i�iklik yap�ld�ktan sonra ve SIMKELMA dozunun titre edilmesinden sonra klinik olarak gerekti�inde serum potasyum d�zeyi izlenmelidir.
Hipokalemi
Hipokalemi g�zlenebilir (Bkz. B�l�m 4.8). Bu gibi durumlarda orta ila �iddetli hipokalemiyi �nlemek i�in idame pozolojisi ba�l��� alt�nda verilen doz titrasyonu gerekli olabilir. �iddetli hipokalemisi olan hastalarda SIMKELMA kesilmeli ve hasta yeniden de�erlendirilmelidir.
QT Uzamas�
Hiperkalemi d�zeltildi�i s�rada, serum potasyum konsantrasyonundaki d�����n fizyolojik sonucu olarak QT aral���nda bir uzama g�zlenebilir.
X-���nlar� ile etkile�im riski
Sodyum zirkonyum siklosilikat, X-���nlar�n� ge�irmeyebilir. E�er hasta bat�n r�ntgeni �ektiriyorsa, radyologlar�n bu durumu g�z �n�nde bulundurmas� gerekmektedir.
�ntestinal perforasyon
SIMKELMA kullan�m� ile intestinal perforasyon riski halihaz�rda bilinmemektedir. SIMKELMA ile herhangi bir intestinal perforasyon olay� bildirilmemi�tir. Gastrointestinal sistemde etki g�steren polimerler ile intestinal perforasyon bildirilmi� oldu�undan, intestinal perforasyonun bulgu ve belirtilerine kar�� �zel dikkat g�sterilmesi gerekmektedir.
Sodyum i�eri�i
Bu t�bbi �r�n her 5g'l�k dozunda 400 mg sodyum ihtiva eder. Bu durum, kontroll� sodyum diyetinde olan hastalar i�in g�z �n�nde bulundurulmal�d�r.
Klinik verilerin k�s�tlamalar�
�iddetli hiperkalemi
Serum potasyum konsantrasyonlar� 6,5 mmol/L'nin �zerinde olan hastalarda s�n�rl� deneyim mevcuttur.
Uzun s�reli maruziyet
SIMKELMA ile klinik �al��malar bir y�ldan daha uzun s�reli maruziyeti i�ermemektedir.
4.5. Di�er t�bbi �r�nler ile etkile�imler ve di�er etkile�im �ekilleri
Di�er t�bbi �r�nlerin sodyum zirkonyum siklosilikat �zerindeki etkisi
Sodyum zirkonyum siklosilikat v�cut taraf�ndan emilmedi�inden veya metabolize edilmedi�inden, di�er t�bbi �r�nlerin sodyum zirkonyum siklosilikat�n farmakolojik aktivitesi �zerinde beklenen bir etkisi bulunmamaktad�r.
Sodyum zirkonyum siklosilikat�n di�er t�bbi �r�nler �zerindeki etkisi
Sodyum zirkonyum siklosilikat v�cut taraf�ndan emilmedi�inden veya metabolize edilmedi�inden ve di�er t�bbi �r�nlere anlaml� d�zeyde ba�lanmad���ndan, di�er t�bbi �r�nlerin �zerinde s�n�rl� etkileri bulunmaktad�r.Sodyumzirkonyumsiklosilikat, hidrojen iyonlar�n�
biyoyararlan�m� pH de�erine ba�l� t�bbi �r�nlerin ��z�n�rl���nde ve emilim kinetiklerinde de�i�ikliklere yol a�abilmektedir. Sa�l�kl� g�n�ll�lerde y�r�t�len bir klinik ila�-ila� etkile�imi �al��mas�nda sodyum zirkonyum siklosilikat ile amlodipin, klopidogrel, atorvastatin, furosemid, glipizid, varfarin, losartan veya levotiroksin ile birlikte uygulanmas�, klinik olarak �nemli ila�-ila� etkile�imleri ile sonu�lanmam��. Di�er gastrik asit modifiye ediciler ile dabigatran�n e�zamanl� uygulamas� ile uyumlu �ekilde, sodyum zirkonyum siklosilikat ile birlikte uyguland���nda dabigatran�n Cve EAA de�erleri yakla��k %40 daha d���k bulunmu�tur. Bu t�bbi �r�nlerin hi�biri i�in herhangi bir doz ayarlamas� veya doz uygulama zamanlar�n�n birbirinden ayr�lmas� gerekmemektedir. Fakat sodyum zirkonyum siklosilikat, mide pH'�na ba��ml� biyoyararlan�m� klinik a��dan anlaml� olan oral ila�lardan en az 2 saat �nce veya 2 saat sonra uygulanmal�d�r.
Artm�� mide pH'� ile ila�lar aras�ndaki olas� etkile�imden ka��nmak ad�na sodyum zirkonyum siklosilikattan 2 saat �nce veya 2 saat sonra uygulanmas� gereken t�bbi �r�nlere verilebilecek �rnekler aras�nda azol antifungaller (ketokonazol, itrakonazol ve posakonazol), anti-HIV ila�lar (atazanavir, nelfinavir, indinavir, ritonavir, sakuinavir, raltegravir, ledipasvir ve rilpivirin) ve tirozin kinaz inhibit�rleri (erlotinib, dasatinib ve nilotinib) yer almaktad�r.
Sodyum zirkonyum siklosilikat, pH'a ba��ml� biyoyararlan�m sergilemeyen oral ila�larla doz uygulama zamanlar� aras�nda aral�k b�rak�lmadan e�zamanl� �ekilde uygulanabilir.
Sa�l�kl� g�n�ll�lerde yap�lan ba�ka bir ila�-ila� etkile�im �al��mas�nda, 15g SIMKELMA'n�n 5mg takrolimus ile birlikte uygulanmas� takrolimus EAA ve C'�nda s�ras�yla %37 ve %29 azalma ile sonu�lanm��t�r. Bu nedenle takrolimus SIMKELMA'dan en az 2 saat �nce veya sonra al�nmal�d�r. Ayn� �al��mada, SIMKELMA ve siklosporinin birlikte uygulanmas� klinik olarak anlaml� bir etkile�im g�stermemi�tir.
�zel pop�lasyonlara ili�kin ek bilgiler Pediyatrik pop�lasyon:
Etkile�im �al��malar� sadece yeti�kinlerde yap�lm��t�r.
4.6. Gebelik ve laktasyon
Gebelik Kategorisi: B
�ocuk do�urma potansiyeli bulunan kad�nlar/Do�um kontrol� (Kontrasepsiyon)
�ocuk do�urma potansiyeli olan kad�nlarda SIMKELMA'n�n kullan�m� hakk�nda veri yoktur. Kontrasepsiyona gerek duyulup duyulmad��� bilinmemektedir.
Gebelik d�nemi
SIMKELMA'n�n gebe kad�nlarda kullan�m�na ili�kin veri bulunmamaktad�r. Hayvan �al��malar� �reme toksisitesine ba�l� olarak do�rudan veya dolayl� zararl� etkileri g�stermemektedir (Bkz. B�l�m 5.3). Tedbir ama�l� olarak gebelik s�ras�nda SIMKELMA kullan�lmamal�d�r.
Laktasyon d�nemi
Emzirmekte olan kad�n�n sodyum zirkonyum siklosilikata sistemik maruz kalmas�, ihmal edilebilir d�zeyde oldu�u i�in, emzirilen �ocuk �zerinde herhangi bir etki �ng�r�lmemektedir. SIMKELMA emzirme d�neminde kullan�labilir.
S��anlardaki bir postnatal �al��mada, sodyum zirkonyum siklosilikata maternal maruziyetin postnatal geli�im �zerinde bir etkisi olmam��t�r. Fizikokimyasal �zelliklerine ba�l� olarak sodyum zirkonyum siklosilikat�n sistemik emilimi bulunmad���ndan anne s�t�ne at�lmas� beklenmez.
�reme yetene�i/Fertilite
�lac�n uyguland��� s��anlarda ya da tav�anlarda embriyo-fetal geli�im �zerinde advers etki olmam��t�r.
4.7. Ara� ve makine kullan�m� �zerindeki etkiler
SIMKELMA'n�n ara� ve makine kullan�m� �zerinde etkisi bulunmamakta ya da etkisi ihmal edilebilecek kadar azd�r.
4.8. �stenmeyen etkiler
G�venlilik profilinin �zeti
En yayg�n s�kl�kla bildirilen advers reaksiyonlar hipokalemi (%4,1) ve �dem ile ili�kili olaylar (%5,7)'d�r.
Advers reaksiyonlar�n tablola�t�r�lm�� �zeti
SIMKELMA'n�n g�venlilik profili, 507'si bir y�l boyunca maruz kalan 1760 hastay� i�eren klinik �al��malarda de�erlendirilmi�tir.
Kontroll� �al��malarda tan�mlanan advers reaksiyonlar Tablo 1'de g�sterilmektedir. Advers reaksiyonlar�n s�kl��� i�in a�a��daki standart kullan�lm��t�r: �ok yayg�n (≥1/10); yayg�n (≥1/100 ila <1/10); yayg�n olmayan (≥1/1.000 ila <1/100); seyrek (≥1/10.000 ila <1/1.000); �ok seyrek (<1/10.000), bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor).
Tablo 1. Klinik �al��malardaki advers reaksiyonlar�n listesi
Sistem Organ s�n�f� |
|
Metabolizma ve beslenme hastal�klar� | Yayg�n: Hipokalemi |
Genel bozukluklar ve uygulama b�lgesine ili�kin hastal�klar | Yayg�n: �dem ile ili�kili olaylar |
Se�ili advers reaksiyonlar�n tan�m�
Hipokalemi
Klinik �al��malarda SIMKELMA hastalar�n�n %4,1'i 3,5 mmol/L'nin alt�nda serum potasyum de�erleriyle seyreden ve SIMKELMA dozunda ayarlama veya SIMKELMA'n�n kesilmesiyle d�zelen hipokalemi geli�tirmi�tir.
�dem ile ili�kili olaylar
A��r� s�v� y�k�, s�v� tutulumu, jeneralize �dem, hipervolemi, lokalize �dem, �dem, periferik �dem ve periferik �i�meyi i�eren �dem ile ili�kili olaylar, SIMKELMA kullanan hastalar�n
%5,7'si taraf�ndan bildirilmi�tir. Bu olaylar sadece idame faz�nda g�zlenmi� ve 15 g ile tedavi edilen hastalarda daha yayg�n olarak g�r�lm��t�r. Olaylar�n %53 kadar� di�retik tedavisine ba�lanarak veya di�retik dozunda ayarlama yap�larak kontrol edilmi�, kalan� ise tedavi gerektirmemi�tir.
Uzun s�reli maruziyet
874 g�n�ll�de 1 y�la varan s�reyle SIMKELMA'ya a��k etiketli maruziyetin s�z konusu oldu�u 2 klinik �al��mada ara�t�rmac�lar taraf�ndan �u olaylar ilgili olarak bildirilmi�tir: gastrointestinal olaylar [konstipasyon (%2,9), diyare (%0,9), abdominal a�r�/distansiyon (%0,5), bulant� (%1,6) ve kusma (%0,5)]; a��r� duyarl�l�k reaksiyonlar� [d�k�nt� (%0,3) ve pr�rit (%0,1)]. Bu olaylar hafif ila orta �iddetli bir yap� sergilemi�tir, hi�biri ciddi �eklinde bildirilmemi�tir ve genel olarak hasta tedaviye devam ederken ��z�mlenmi�tir. A��k etiketli �al��ma tasar�m� nedeniyle bu olaylar ile SIMKELMA aras�nda bir nedensel ili�ki kesin olarak ortaya konamamaktad�r.
��pheli advers reaksiyonlar�n raporlanmas�
Ruhsatland�rma sonras� ��pheli ila� advers reaksiyonlar�n�n raporlanmas� b�y�k �nem ta��maktad�r. Raporlama yap�lmas�, ilac�n yarar / risk dengesinin s�rekli olarak izlenmesine olanak sa�lar. Sa�l�k mesle�i mensuplar�n�n herhangi bir ��pheli advers reaksiyonu T�rkiye Farmakovijilans Merkezi (T�FAM)'ne bildirmeleri gerekmektedir (www.titck.gov.tr; e-posta: tufam@titck.gov.tr; tel: 0 800 314 00 08; faks: 0 312 218 35 99).
4.9. Doz a��m� ve tedavisi
Sodyum zirkonyum siklosilikat ile doz a��m� hipokalemiye neden olabilir. Hastalar�n serum potasyum d�zeyleri kontrol edilmeli ve gerekirse potasyum takviyesi yap�lmal�d�r.
5. FARMAKOLOJ�K �ZELL�KLER
5.1. Farmakodinamik �zellikler
Farmakoterap�tik grup: Hiperkalemi ve hiperfosfatemi tedavisinde kullan�lan ila�lar ATC kodu: V03AE10
Etki mekanizmas�:
Sodyum zirkonyum siklosilikat, hidrojen ve sodyum katyonlar�n�n de�i�-toku� de�i�iminde potasyumu tercihli bir �ekilde yakalayan tekd�ze mikrog�zenekli yap�ya sahip absorbe olmayan, polimer olmayan bir inorganik tozdur. Sodyum zirkonyum siklosilikat, in vitro ortamda kalsiyum ve magnezyum gibi di�er katyonlar�n varl���nda dahi potasyum iyonlar� i�in y�ksek d�zeyde se�icidir. Sodyum zirkonyum siklosilikat, gastrointestinal (G�) sistemin tamam�nda potasyumu yakalar ve G� l�mende serbest potasyum konsantrasyonunu d���r�r, b�ylece hiperkalemiyi d�zeltmek i�in serum potasyum d�zeylerini azalt�r ve fe�es ile potasyum at�l�m�n� art�r�r.
Farmakodinamik etkiler
Sodyum zirkonyum siklosilikat, al�m�ndan sonraki 1 saat kadar k�sa bir s�re i�inde serum potasyum konsantrasyonlar�n� d���rmeye ba�lar ve normokalemiye genel olarak 24 ila 48 saat
konsantrasyonlar�n� ya da idrarla sodyum at�l�m�n� etkilemez. Ba�lang�� serum potasyum d�zeyleri ile etkinin b�y�kl��� aras�nda yak�n bir korelasyon vard�r; ba�lang�� serum potasyum d�zeyleri daha y�ksek hastalarda serum potasyum d�zeylerindeki azalmalar daha fazla olmaktad�r. Serum potasyum konsantrasyonunda azalman�n bir sonucu olarak idrarla potasyum at�l�m�nda bir d���� meydana gelir. D�rt g�n s�reyle g�nde bir kez SIMKELMA 5 g veya 10 g verilen sa�l�kl� g�n�ll�lerle ger�ekle�tirilen bir �al��mada, serum potasyum konsantrasyonu ve idrarla toplam potasyum at�l�m�ndaki doza ba��ml� azalmaya, fe�es ile potasyum at�l�m�nda ortalama art��lar e�lik etmi�tir. �drarla sodyum at�l�m�nda istatistiksel olarak anlaml� de�i�iklikler g�zlenmemi�tir.
Sodyum zirkonyum siklosilikat�n a� veya tok karn�na uyguland��� durumlarda farmakokineti�ini inceleme ama�l� �al��malar ger�ekle�tirilmemi�tir.
Sodyum zirkonyum siklosilikat�n ayr�ca in vitro ve in vivo ko�ullarda amonyumu ba�lad���, b�ylelikle amonyumu uzakla�t�r�p serum bikarbonat d�zeylerini art�rd��� g�sterilmi�tir. SIMKELMA ile tedavi edilen hastalar bikarbonat d�zeyinde g�nde bir kez 5 g dozunda 1,1 mmol/L, g�nde bir kez 10 g dozunda 2,3 mmol/L ve g�nde bir kez 15 g dozunda 2,6 mmol/L art�� ya�am��, plasebo uygulanan hastalarda ise ortalama art�� 0,6 mmol/L olmu�tur. Renin ve aldosteronu etkileyen di�er fakt�rlerin kontrol edilmedi�i bir ortamda SIMKELMA, plasebo grubu (+%14) ile kar��la�t�r�ld���nda, ortalama serum aldosteron d�zeylerinde dozdan ba��ms�z bir de�i�im sa�lam��t�r (aral�k: -%30 ila -%31). Ancak bununla ili�kili olarak sistolik ve diyastolik kan bas�nc�nda herhangi bir etki g�zlenmemi�tir.
Ek olarak, plasebo (0,8 mg/dL) ve d���k doz sodyum zirkonyum siklosilikat (0,3 mg/dL) gruplar�ndaki k���k ortalama art��lar ile kar��la�t�r�ld���nda g�nde �� kez 5 g (1,1 mg/dL) ve
10 g (2,0 mg/dL) gruplar�nda serum �re nitrojen (B�N) de�erlerinde ortalama d����ler g�zlenmi�tir.
Klinik etkililik ve g�venlilik:
SIMKELMA'n�n potasyum d���r�c� etkisi, hiperkalemisi olan hastalarda y�r�t�len randomize, �ift k�r, plasebo kontroll� �� �al��mada g�sterilmi�tir. �� �al��ma 48 saatlik bir periyot boyunca SIMKELMA'n�n hiperkalemiyi d�zeltmedeki ilk etkisini test etmi� ve iki �al��ma ayr�ca elde edilen normokalemi etkisinin idamesini test etmi�tir. �dame �al��malar�na, kronik b�brek hastal��� (% 58), kalp yetmezli�i (% 10), diabetes mellitus (% 62) ve RAAS inhibit�r tedavisi (% 68) olan hastalar dahil edilmi�tir. Ek olarak iki a��k etiketli idame �al��mas�nda SIMKELMA'n�n uzun s�reli g�venlili�i test edilmi�tir. Bu be� �al��mada SIMKELMA dozlar� verilen 1760 hasta yer alm��t�r; bu hastalar�n 507'si ilaca en az 360 g�n maruz kalm��t�r. Ek olarak, SIMKELMA'n�n etkilili�i ve g�venlili�i, 8 hafta boyunca SIMKELMA dozlar� alan hiperkalemili 196 kronik hemodiyaliz hastas� ile y�r�t�len �ift k�r, plasebo kontroll� bir �al��mada incelenmi�tir. Bu �al��malarda SIMKELMA, altta yatan hiperkalemi sebebi, ya�, cinsiyet, �rk, komorbid hastal�k ve e�zamanl� RAAS inhibit�rleri kullan�m�ndan ba��ms�z olarak serum potasyumu d�zeyini d���rm�� ve normal serum potasyum d�zeylerini korumu�tur. Herhangi bir diyet k�s�tlamas� getirilmemi� olup, hastalara �zel bir de�i�iklik olmaks�z�n her zamanki diyetlerine devam etmeleri s�ylenmi�tir.
![]()
�ki fazl�, plasebo kontroll� d�zeltme ve idame kullan�m� �al��mas�
Hiperkalemisi (5 ila ≤6,5 mmol/L, ba�lang��taki potasyum ortalamas� 5,3 mmol/L) olan ve kronik b�brek hastal���, kalp yetmezli�i ve diyabeti olan ve RAAS inhibit�r tedavisinde olan hastalar�n dahil oldu�u 753 hastada (ortalama ya� 66 y�l, aral�k 22 ila 93) y�r�t�len 2 k�s�ml�, �ift k�r, randomize, plasebo kontroll� bir �al��mad�r.
D�zeltme faz� s�ras�nda hastalar, ilk 48 saat s�reyle g�nde �� kez uygulanan SIMKELMA (1,25 g, 2,5 g, 5 g veya 10 g) ya da plasebo almak �zere randomize edilmi�tir (Tablo 2).
Tablo 2. D�zeltme faz� (�al��ma 1): 48 saat SIMKELMA sonras�nda normokalemik olgular�n y�zdesi
SIMKELMA dozu (g�nde �� kez) | |||||
| Plasebo | 1,25 g | 2,5 g | 5 g | 10 g |
N | 158 | 154 | 141 | 157 | 143 |
Ba�lang�� serum potasyum, mmol/L | 5,3 | 5,4 | 5,4 | 5,3 | 5,3 |
48 saatte normokalemik, % | 48 | 51 | 68 | 78 | 86 |
Plaseboya kar�� p de�eri |
| AD | <0,001 | <0,001 | <0,001 |
AD: anlaml� de�il
G�nde �� kez uygulanan SIMKELMA 10 g, 48 saatte serum potasyum d�zeyini 0,7 mmol/L d���rm��t�r (plaseboya kar�� p<0,001); ilk dozdan 1 saat sonra istatistiksel olarak anlaml� %14 potasyum d����� g�zlenmi�tir. Ba�lang��ta potasyum d�zeyleri daha y�ksek olan hastalar�n SIMKELMA'ya yan�t� daha fazla olmu�tur. Tedavi �ncesi potasyum d�zeyleri 5,5 mmol/L'nin �zerinde olan hastalar (ortalama ba�lang�� de�eri 5,8 mmol/L) 48 saatte 1,1 mmol/L'lik ortalama bir d���� elde ederken ba�lang��taki potasyum d�zeyleri 5,3 mmol/L veya bu de�erin alt�nda olanlarda ortalama d����, en y�ksek dozda 0,6 mmol/L olmu�tur.
D�zeltme faz� s�ras�nda SIMKELMA ald�ktan sonra normokalemik duruma ge�en hastalar, g�nde bir kez plasebo veya d�zeltme faz� s�ras�nda g�nde �� kez ald�klar� ile ayn� doz d�zeyinde g�nde bir kez SIMKELMA almak �zere tekrar randomize edilmi�tir (Tablo 3).
Tablo 3. �dame faz� (12 g�n, �al��ma 1): Ortalama normokalemik g�n say�s�
�dame faz� tedavisi (g�nde bir kez) | |||||
| Plasebo |
| SIMKELMA | Plaseboya p de�eri | |
D�zeltme faz� SIMKELMA dozu | n | G�n | n | G�n |
|
1,25 g g�nde �� kez | 41 | 7,6 | 49 | 7,2 | AD |
2,5 g g�nde �� kez | 46 | 6,2 | 54 | 8,6 | 0,008 |
5 g g�nde �� kez | 68 | 6,0 | 64 | 9,0 | 0,001 |
10 g g�nde �� kez | 61 | 8,2 | 63 | 10,2 | 0,005 |
AD: anlaml� de�il
SIMKELMA'n�n art�k uygulanmad��� idame periyodunun sonunda, ortalama potasyum d�zeyleri, ba�lang�� d�zeylerineyak�nbirseviyeyey�kselmi�tir.
![]()
Ek bir a��k etiket fazl�, �ok merkezli, plasebo kontroll� idame �al��mas�
�al��man�n d�zeltme faz�nda, hiperkalemisi olan 258 hasta (ba�lang�� ortalamas� 5,6, aral�k 4,1
- 7,2 mmol/L), 48 saat s�reyle g�nde �� kez 10 g SIMKELMA alm��t�r. SIMKELMA'n�n
10 g'l�k ilk dozundan 1 saat sonra potasyum d�zeylerinde d����ler g�zlenmi�tir. Normokalemiye kadar ge�en medyan s�re 2,2 saat olmu�, hastalar�n %66's� 24 saatte ve %88'i 48 saatte normokalemiye ula�m��t�r. Hiperkalemisi daha �iddetli olan hastalarda yan�tlar daha b�y�k olmu�tur; ba�lang�� serum potasyum d�zeyleri <5,5, 5,5-5,9 ve ≥6 mmol/L olan hastalarda serum potasyum d�zeyi s�ras�yla 0,8, 1,2 ve 1,5 mmol/L d��m��t�r.
Normokalemiye ula�an hastalar (potasyum d�zeyleri 3,5 ile 5 mmol/L aras�nda) �ift k�r olarak 28 g�n s�reyle (�ift k�r randomize doz azaltma faz�) g�nde bir kez uygulanan (�ift k�r randomize doz azaltma faz�) SIMKELMA'n�n �� dozundan birine [5 g (n=45), 10 g (n=51) veya 15 g (n=56)] ya da plaseboya (n=85) randomize edilmi�tir.
�al��mada G�n 8'den G�n 29'a (�� haftal�k periyot) ortalama serum potasyum d�zeyi
<5,1 mmol/L olan olgular�n oran�, plasebo (%46) ile kar��la�t�r�ld���nda, SIMKELMA'n�n g�nde bir kez 5 g, 10 g ve 15 g dozlar�nda (s�ras�yla %80, %90 ve %94) daha y�ksek bulunmu�tur. Serum potasyum de�erinde s�ras�yla 0,44 mmol/L, 0,77 mmol/L, 1,10 mmol/L ve 1,19 mmol/L ortalama d���� olmu�tur ve normokalemik kalan olgular�n oran�, g�nde bir doz SIMKELMA 5 g, 10 g, 15 g ve plasebo gruplar�nda s�ras�yla %71, %76, %85 ve %48 olmu�tur.
SIMKELMA titrasyonu (a��k etiketli) ile idame faz� sonu�lar�: 123 hasta 11 ayl�k a��k etiketli faza girmi�tir. Serum potasyum de�erini etkileyebilen di�er fakt�rlere bak�lmaks�z�n ortalama serum potasyum d�zeyi <5,1 mmol/L olan olgular�n oran� %88 ve ortalama serum potasyum d�zeyi 4,66 mmol/L idi. Serum potasyum d�zeyi 3,5 mmol/L'nin alt�nda olan olgular�n oran�
%1'in alt�nda; serum potasyum d�zeyi 3,5 ile 5,1 mmol/L aras�nda olan olgular�n oran� %77 veya serum potasyum seviyesi 3,5 ile 5,5 mmol/L aras� olan olgular�n oran� %93 olmu�tur. �al��ma tamamland���nda (G�n 365) tedavi kesilmi�tir.
�dame faz�nda relapsa kadar ge�en zamana ili�kin Kaplan-Meier tahminleri, relapsa kadar ge�en zamanda doza ba��ml�l�k oldu�unu g�stermi�, 5 g dozu i�in medyan s�re ba�lang��taki serum potasyum d�zeylerine ba�l� olarak 4 ila 21 g�n aras�nda de�i�mi�tir. Serum potasyum d�zeyinin periyodik olarak izlenmesi ve SIMKELMA dozunun B�l�m Pozoloji ve uygulama �ekli ba�l��� alt�nda belirtildi�i �ekilde titre edilmesi gerekmektedir.
�ekil 1 �al��man�n d�zeltme ve idame fazlar�ndaki ortalama serum potasyumu g�stermektedir.
�ekil 1. D�zeltme ve idame fazlar� (�al��ma 2): zaman i�inde g�zlenen ortalama serum potasyum d�zeyleri ve %95 CI de�erleri
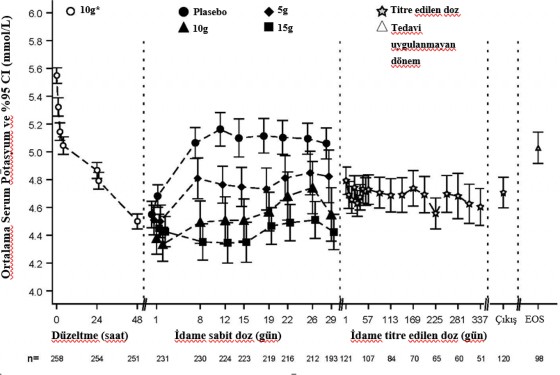
��k��=Son Dozun uygulanmas�n� takip eden 1 g�n i�indeki Son Ziyaret, EOS=�al��ma Sonu (Son Dozdan sonra 7 g�n +/- 1 g�n)
*G�nde �� defa
�al��ma 3
Hiperkalemili kronik b�brek hastal��� olan hastalarda bir �al��ma
Bu �al��ma, ba�lang�� eGFR'si 30 ila 60 ml/dk/1,73 m ve hiperkalemisi (ba�lang�� serum potasyum de�eri 5,2 mmol/L, aral�k: 4,6 - 6 mmol/L) olan 90 hastada (60 SIMKELMA hastas�; 30 kontrol) y�r�t�len �ift k�r, plasebo kontroll� bir doz y�kseltme �al��mas�d�r. Hastalar, iki ila d�rt g�n s�reyle g�nde �� kez yemek ile birlikte uygulanan SIMKELMA'n�n y�kselen dozlar�na (0,3 g, 3 g ve 10 g) veya plasebo kollar�na randomize edilmi�tir. Birincil sonlan�m noktas� tedavinin ilk 2 g�n� boyunca serum potasyum d�zeyinde ba�lang�ca g�re de�i�im oran� olmu�tur. �al��ma, plasebo ile kar��la�t�r�ld���nda SIMKELMA'n�n 3 g ve 10 g dozlar�nda birincil etkililik sonlan�m noktas�na ula�m��t�r. SIMKELMA 10 g dozunda ve 3 g dozunda s�ras�yla 0,92 mmol/L ve 0,43 mmol/L'lik ortalama maksimum d����ler ile sonu�lanm��t�r. Yirmi d�rt saatlik idrar �rnekleri SIMKELMA'n�n idrarla potasyum at�l�m�n� ba�lang�ca k�yasla d���rd���n� g�stermi�tir; plasebo 8,9 mmol/24 saat ile kar��la�t�r�ld���nda 15,8 mmol/24 saat, (p <0,001). Sodyum at�l�m� plaseboya g�re de�i�memi�tir (plasebo 36,9 mmol/24 saat (AD) ile kar��la�t�r�ld���nda 10 g i�in 25,4 mmol/24 saat).
�al��ma 4
�ki fazl�, �ok merkezli, �ok dozlu, a��k etiketli bir g�venlik ve etkinlik �al��mas� SIMKELMA'n�n uzun s�reli (12 aya kadar) etkileri bu �al��mada hiperkalemili 751 g�n�ll� �zerinde ara�t�r�lm��t�r (ba�lang�� ortalamas� 5,59 mmol/L; aral�k 4,3 – 7,6 mmol/L). Komorbid durumlar kronik b�brek hastal��� (%65), diabetes mellitus (%64), kalp yetmezli�i (%15) ve hipertansiyonu i�ermi�tir (%83). G�n�ll�lerin s�ras�yla %51 ve 70'inde di�retik ve RAAS inhibit�r� kullan�m� bildirilmi�tir. D�zeltme faz� s�ras�nda en az 24 saat s�resince ve 72 saate varan s�reyle g�nde �� defa 10 g SIMKELMA uygulanm��t�r. Normokaleminin (3,5 – 5,0 mmol/L, s�n�r de�erleri dahil) 72 saat i�inde sa�land��� g�n�ll�ler �al��man�n idame faz�na girmi�tir. �dame faz�ndaki t�m g�n�ll�ler g�nde bir defa 5 g �eklinde bir ba�lang�� dozuyla SIMKELMA alm��t�r ve bu doz g�nde bir defa 5 g'lik art��larla y�kseltilebilmi�tir (g�nde bir defa maksimum 15 g'ye kadar) veya titrasyon rejimine g�re azalt�labilmi�tir (iki g�nde bir uygulanan minimum 5 g'ye kadar).
D�zeltme faz� dozunun uygulamas�ndan 24, 48 ve 72 saat sonra g�n�ll�lerin 494/748 (%66), 563/748 (%75) ve 583/748'inde (%78) normokalemi sa�lanm��t�r ve serum potasyumdaki ortalama azalma 24 (n=748), 48 (n=104) ve 72 (n=28) saatte s�ras�yla 0,81 mmol/L, 1,02 mmol/L ve 1,10 mmol/L olmu�tur. Normokaleminin ba�lang��taki potasyum konsantrasyonuna ba�l� oldu�u g�r�lm��t�r ve ba�lang��ta en y�ksek serum potasyum konsantrasyonlar�na sahip olan g�n�ll�lerin �al��ma ilac�na ba�lad�ktan sonra en belirgin azalmay� ya�ad��� fakat normokalemi sa�lanan g�n�ll�lerin oran�n�n bu grupta en d���k oldu�u g�zlenmi�tir. Y�z yirmi alt� hastan�n ba�lang�� serum potasyum de�erinin ≥ 6,0 mmol/L oldu�u belirlenmi�tir (ba�lang��taki ortalama potasyum 6,28 mmol/L). Bu g�n�ll�lerde ortalama azalman�n d�zeltme faz�n�n sonunda 1,37 mmol/L oldu�u tespit edilmi�tir.
Tablo 4. D�zeltme faz� (�al��ma 4): d�zeltme faz�ndaki �al��ma g�n�ne g�re serum potasyum konsantrasyonlar� 3,5 ile 5,0 mmol/L aras�nda (s�n�r de�erleri dahil) veya 3,5 ve 5,5 mmol/L (s�n�r de�erleri dahil) aras�nda olan g�n�ll�lerin oran� – ITT pop�lasyon
D�zeltme Faz� (DF) | G�nde �� defa 10 g SIMKELMA (N=749) | |||||
Serum potasyum 3,5 ila 5,0 mmol/L (s�n�r de�erleri dahil) | Serum potasyum 3,5 ila 5,5 mmol/L (s�n�r de�erleri dahil) | |||||
n/N | Oran | %95 CI | n/N | Oran | %95 CI | |
DF 24 saat | 494/748 | 0,660 | 0,625, 0,694 | 692/748 | 0,925 | 0,904, 0,943 |
DF 48 saat | 563/748 | 0,753 | 0,720, 0,783 | 732/748 | 0,979 | 0,965, 0,988 |
DF 72 saat / DF Son | 583/748 | 0,779 | 0,748, 0,809 | 738/748 | 0,987 | 0,976, 0,994 |
Not: Bir g�n�ll�n�n doz sonras� de�eri son dozu takip eden 1 g�nden daha sonras�na aitti. Dolay�s�yla bu g�n�ll� D�zeltme Faz�n�n ITT Pop�lasyonu i�in uygundu; fakat ilgili zaman noktas� analize dahil edilmedi.
Hastalar ilac� almaya devam ederken normokalemi korunmu�tur ve ortalama serum potasyum de�eri ilac�n b�rak�lmas�n� takiben artm��t�r. Ba�lang��ta RAAS inhibit�rleri kullanan hastalar�n
%89'u RAAS inhibit�r� tedavisini b�rakmam��t�r, %74'� idame faz� s�ras�nda ayn� dozda kalabilmi�tir, ba�lang��ta RAAS inhibit�rleri kullanmayanlar�n ise %14'� bu tedaviye ba�layabilmi�tir. �dame faz� s�ras�nda g�n�ll�lerin %75,6's� RAAS inhibit�r� kullan�m�na ra�men normokalemiyi korumu�tur.
�ekil 2'de �al��man�n d�zeltme ve idame fazlar�ndaki ortalama serum potasyum g�sterilmektedir.

%95 CI ile serum potasyum (mmol/L)
�ekil 2: 12 ayl�k a��k etiketli �al��man�n (�al��ma 4) d�zeltme ve idame fazlar� – %95 CI ile zaman i�indeki ortalama serum potasyum
CPBL=D�zeltme Faz� Ba�lang�c�, MPBL=�dame Faz� Ba�lang�c�
��k��=Son Dozu Takip Eden 1 G�n ��indeki Son Ziyaret, EOS=�al��ma Sonu (Son Dozun Ard�ndan 7 g�n +/- 1 g�n)
�al��ma 5
Kronik hemodiyaliz uygulanan hastalar �zerinde ger�ekle�tirilen randomize, �ift k�r, plasebo kontroll� �al��ma
Bu �al��mada son d�nem b�brek hastal��� g�r�len, en az 3 ayd�r stabil diyaliz g�ren ve persistan diyaliz �ncesi hiperkalemi sergileyen 196 hasta (ortalama ya� 58, aral�k 20 ila 86 ya�), diyaliz uygulanmayan g�nlerde g�nde bir defa 5 g SIMKELMA veya plasebo almak �zere randomize edilmi�tir. Randomizasyon s�ras�ndaki ortalama serum potasyum seviyelerinin SIMKELMA grubunda 5,8 mmol/L (aral�k 4,2-7,3 mmol/L), plasebo grubunda ise 5,9 mmol/L (aral�k 4,2– 7,3 mmol/L) oldu�u belirlenmi�tir. Doz ayarlama periyodu s�ras�nda (ilk 4 hafta) 4,0-5,0 mmol/L aras� bir diyaliz �ncesi serum potasyum seviyesine ula�mak amac�yla doz, LIDI sonras� serum potasyum �l��m�ne g�re, haftal�k olarak 5 g'l�k art��larla ayarlanabilmi�tir. Doz ayarlama periyodunun sonunda ula��lan doz takip eden 4 haftal�k de�erlendirme periyodu s�resince devam ettirilmi�tir. Doz ayarlama periyodunun sonunda hastalar�n %37, %43 ve
%19'unun 5 g, 10 g ve 15 g SIMKELMA almakta oldu�u belirlenmi�tir. De�erlendirme periyodu s�ras�nda kurtarma tedavisi g�rmeyenler ve LIDI sonras� 4 diyaliz tedavisinin en az 3'�nde diyaliz �ncesi serum potasyum d�zeyinin 4,0 ile 5,0 mmol/L aras�nda korundu�u g�n�ll�ler �eklinde tan�mlanan yan�t verenlerin oran� SIMKELMA grubunda %41, plasebo grubunda ise %1 olmu�tur (p<0,001)(Bkz.�ekil 3).
LIDI sonras� post-hoc analizlerde de�erlendirme periyodu s�ras�nda hastalar�n serum potasyum seviyesinin 4,0 ile 5,0 mmol/L aras�nda oldu�u durumlar�n say�s� SIMKELMA grubunda daha y�ksek olmu�tur. SIMKELMA grubunda 4 ziyaretin t�m�nde de�erleri bu aral�kta olan hastalar�n oran�n�n %24 oldu�u belirlenmi�tir, plasebo grubunda ise bu oran %0 olmu�tur. Post- hoc analiz, de�erlendirme periyodu s�ras�nda LIDI sonras� 4 diyaliz tedavisinin en az 3'�nde serum potasyum d�zeyinin 3,5 ile 5,5 mmol/L aras�nda korundu�u hastalar�n oran�n�n SIMKELMA grubunda %70, plasebo grubunda ise %21 oldu�unu g�stermi�tir.
Tedavinin sonunda, ortalama diyaliz sonras� serum potasyum seviyesi SIMKELMA grubunda 3,6 mmol/L (aral�k 2,6-5,7 mmol/L), plasebo grubunda ise 3,9 mmol/L (aral�k 2,2-7,3 mmol/L) olmu�tur. Diyaliz aras� kilo art��� (IDWG) a��s�ndan SIMKELMA ve plasebo gruplar� aras�nda fark g�zlenmemi�tir. IDWG, diyaliz �ncesi kilo eksi �nceki diyaliz seans�ndaki diyaliz sonras� kilo �eklinde tan�mlanm��t�r ve LIDI sonras�nda �l��lm��t�r.
�ekil 3: Kronik diyaliz uygulanmakta olan hastalarda ortalama diyaliz �ncesi serum potasyum seviyelerinin zaman i�indeki seyri
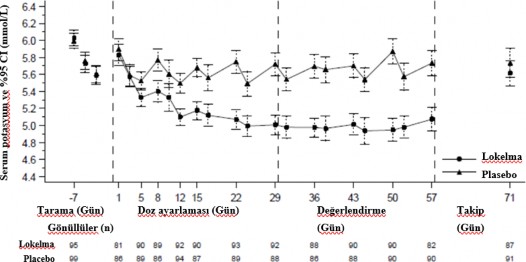
G�sterilen hata �ubuklar� %95 g�ven aral�klar�na kar��l�k gelmektedir.
n = Belirli bir ziyarette potasyum �l��mleri eksik olmayan hastalar�n say�s�.
5.2. Farmakokinetik �zellikler
Emilim:
Klinik �al��malar, sistemik emilimi olmad���n� g�stermi�tir. S��anlardaki bir in vivo k�tle dengesi �al��mas�, sodyum zirkonyum siklosilikat�n herhangi bir sistematik emilim kan�t� olmadan fe�este tespit edildi�ini g�stermi�tir. Bu fakt�rler ve ��z�n�r olmamas� nedeniyle, sitokrom P450 (CYP450) enzimleri veya ta��y�c� aktivitesi �zerindeki etkisini de�erlendirmek i�in in vivo veya in vitro �al��ma ger�ekle�tirilmemi�tir.
Da��l�m:
Sistemik da��l�m� bulunmamaktad�r.
Biyotransformasyon:
Sodyum zirkonyum siklosilikat, enzimatik olarak metabolize olmayan inorganik, ��z�nmeyen bir bile�iktir.
Eliminasyon:
Sodyum zirkonyum siklosilikat fe�es yoluyla elimine edilir.
Do�rusall�k/ do�rusal olmayan durum:
Uygulanabilir de�ildir.
5.3. Klinik �ncesi g�venlilik verileri
Klinik d��� veriler; g�venlilik farmakolojisi, tekrarl� doz toksisitesi, genotoksisite, karsinojenik potansiyel, �reme ve geli�im toksisitesi standart �al��malar�na dayal� olarak insanlar i�in herhangi bir �zel tehlike ortaya koymamaktad�r.
6. FARMAS�T�K �ZELL�KLER
6.1. Yard�mc� maddelerin listesi
Bulunmamaktad�r.
6.2. Ge�imsizlikler
Ge�erli de�ildir.
6.3. Raf �mr�
36 ay
6.4. Saklamaya y�nelik �zel tedbirler
30°C alt�ndaki oda s�cakl���nda saklay�n�z.
6.5. Ambalaj�n niteli�i ve i�eri�i
3-katmanl� al�minyum folyo laminattan (PET/al�minyum/LLDPE) yap�lm�� sa�eler i�inde 5 g toz.
Ambalaj b�y�kl�kl���: 11 veya 28 sa�e
6.6. Be�eri t�bbi �r�nden arta kalan maddelerin imhas� ve di�er �zel �nlemler
Tek dozluk sa�enin t�m i�eri�i, yakla��k 45 mL su i�eren bir su barda��na bo�alt�lmal� ve iyice kar��t�r�lmal�d�r. Toz ��z�lmeyecektir. Tad� olmayan s�v�, hen�z bulan�kken i�ilmelidir. E�er toz ��kerse su yeniden kar��t�r�lmal�d�r. T�m i�eri�in i�ildi�inden emin olunmal�d�r.
Kullan�lmam�� olan �r�nler ya da at�k materyaller “T�bbi At�klar�n Kontrol� Y�netmeli�i” ve “Ambalaj At�klar�n�n Kontrol� Y�netmelik”lerine uygun olarak imha edilmelidir.
 Kolon, Rektum yada Ba��rsak Kanseri
Ba��rsak kanseri kolon veya rektumda
(arka ge�it) herhangi bir b�lgede ortaya ��kabilir.Kolorektal kanser erken safhalarda te�his edilmesi halinde daha kolay ve daha ba�ar�l� bir
�ekilde tedavi edilir.
Kolon, Rektum yada Ba��rsak Kanseri
Ba��rsak kanseri kolon veya rektumda
(arka ge�it) herhangi bir b�lgede ortaya ��kabilir.Kolorektal kanser erken safhalarda te�his edilmesi halinde daha kolay ve daha ba�ar�l� bir
�ekilde tedavi edilir. |
 Travma Sonras� Bunal�m�
Travmatik bir olay, g�nl�k ola�an olaylar�n d���nda olan ve ki�iyi derinden
rahats�z eden bir olayd�r.Bir�ok olay b�yle bir etki g�sterebilir.
Travma Sonras� Bunal�m�
Travmatik bir olay, g�nl�k ola�an olaylar�n d���nda olan ve ki�iyi derinden
rahats�z eden bir olayd�r.Bir�ok olay b�yle bir etki g�sterebilir. |
 |
S�rt A�r�s� S�rt a�r�s� birden bire ortaya ��k�p �iddetli (akut) olabilir veya zamanla geli�ip daha uzun s�reli sorunlara (kronik) neden olabilir. |
 |
Mesane Kanseri Mesane kanseri her zaman mukozada ba�lar. Erken safhalarda bu tabakada s�n�rl� kal�r ve h�cre i�indeki karsinom olarak nitelendirilir. |
 |
�nme �nme, beynin hasar g�rmesinin sonucudur. Bu hasar, beynin bir k�sm�ndaki ya bir kanama ya da akut kan eksikli�i nedeniyle o k�sm�n ge�ici ya da kal�c� olarak i�levini yapamamas�na yol a�ar. |
�LA� GENEL B�LG�LER�
AstraZeneca T�rkiye �la� Sanayi ve Ticaret Ltd.�ti.
| Sat�� Fiyat� | 8271.03 TL [ 3 Aug 2026 ] |
| �nceki Sat�� Fiyat� | 8271.03 TL [ 27 Jul 2026 ] |
| Original / Jenerik | Original �la� |
| Re�ete Durumu | Normal Re�eteli bir ila�d�r. |
| Barkodu | 8699786250031 |
| Etkin Madde | Sodyum Zirkonyum Siklosilikat |
| ATC Kodu | V03AE10 |
| Birim Miktar | 5 |
| Birim Cinsi | G |
| Ambalaj Miktar� | 11 |
| �e�itli �la�lar > Di�er T�m �la�lar |
| �thal ( ref. �lke : Abd ) ve Be�eri bir ila�d�r. |