PIRFA 200 mg 252 film tablet K�sa �r�n Bilgisi
{ Pirfenidon }
1. BE�ER� TIBB� �R�N�N ADI
P�RFA® 200 mg Film Kapl� Tablet
2. KAL�TAT�F VE KANT�TAT�F B�LE��M
Etkin madde
Her bir tablet, 200 mg pirfenidon i�erir.
Yard�mc� maddeler
Laktoz monohidrat (inek s�t�) 56,00 mg Yard�mc� maddeler i�in b�l�m 6.1'e bak�n�z.
3. FARMAS�T�K FORMU
Film kapl� tablet
A��k sar� renkli, oval film tablet
4. KL�N�K �ZELL�KLER
4.1. Terap�tik endikasyonlar
P�RFA®, yeti�kinlerde hafif ve orta derecede �diyopatik Pulmoner Fibrozis (�PF)'in tedavisinde endikedir.
4.2. Pozoloji ve uygulama �ekli
P�RFA doz �emas� | |
Ba�lang�� dozu (800 mg/g�n) | g�nde 4 defa; 1 adet 200 mg/tablet |
�dameye ge�i� dozu (1600 mg/g�n) | g�nde 4 defa; 2 adet 200 mg/tablet |
�dame doz (2400 mg/g�n) | g�nde 4 defa; 3 adet 200 mg/tablet |
P�RFA® tedavisi, �PF tan� ve tedavisinde uzman hekimler taraf�ndan y�r�t�lmelidir. Yeti�kinler i�in;
P�RFA® i�in �nerilen ba�lang�� dozu, toplam doz g�nde 800 mg/g�n olacak �ekilde yemeklerle birlikte al�nan g�nde 4 defa 200 mg/tablettir. Ayr�ca doz, semptomlara ba�l� olarak uygun �ekilde art�r�labilir.
Tabletler oral yolla b�t�n halinde, bir miktar su ile al�nmal�d�r. Bulant�, sersemlik gibi yan etkilerin olu�ma riskinin azalt�lmas� i�in mutlaka yemeklerden sonra kullan�lmal�d�r.
2400 mg/g�n �zerindeki dozlar hi�bir hastada kullan�lmamal�d�r.
14 ard���k g�n veya daha fazla s�reyle pirfenidon tedavisi almayan hastalar, ba�lang�� dozundan ba�layarak, kademeli olarak ve g�zlem alt�nda, �nerilen g�nl�k doza kadar s�ren tedaviye yeniden ba�lamal�d�r.
14 ard���k g�nden daha az bir zaman i�in tedavi kesilirse, titrasyon yap�lmaks�z�n �nceden �nerilen g�nl�k dozdan tedaviye devam edilebilir.
Doz Ayarlamas� ve G�venli Kullan�m i�in �neriler
Gastrointestinal olaylar: Gastrointestinal yan etkilerden dolay� tedaviye intolerans g�steren hastalara, ilac� yiyeceklerle birlikte almas� gerekti�i hat�rlat�lmal�d�r. E�er semptomlar s�rerse, P�RFA® dozu 1-2 tablet azalt�labilir ve g�nde 4 defa yemeklerle birlikte tolere edilebilen �nerilen g�nl�k doza yeniden art�r�labilir. E�er semptomlar devam ederse, hasta semptomlar� gidermek ad�na tedaviye 1-2 hafta ara verme konusunda bilgilendirilmelidir.
Fotosensitivite reaksiyonu veya k�zar�kl�k: Hafif veya orta derecede fotosensitivite reaksiyonu veya k�zar�kl�k/d�k�nt� ya�ayan hastalara g�ne� koruyucu krem kullan�m� �nerilmeli ve g�ne�e maruziyeti �nleme bilgisi verilmelidir. P�RFA® dozu g�nde 800 mg'a azalt�labilir (g�nde 4 defa 200 mg'l�k 1 tablet). E�er k�zar�kl�k 7 g�n sonra da devam ederse, P�RFA® 15 g�n i�in kullan�lmamal�, doz art�rma periyoduyla ayn� olacak �ekilde �nerilen g�nl�k doza yeniden art�r�lmal�d�r.
A��r fotosensitivite reaksiyonu veya k�zar�kl�k g�zlenen hastalar tedaviyi kesmek ve medikal destek alma konusunda bilgilendirilmelidirler. K�zar�kl�k ge�ti�inde, P�RFA® yeniden ba�lanabilir ve hekimin y�nlendirmesine g�re �nerilen g�nl�k doza yeniden art�r�labilir.
Uygulama �ekli:
P�RFA® oral uygulama i�indir. P�RFA® bir bardak su ile yutulmal�d�r, bulant�y� �nlemek i�in yiyeceklerle birlikte al�nmal�d�r.
�zel pop�lasyonlara ili�kin ek bilgiler:
Karaci�er yetmezli�i:
Hafif ve orta derecede hepatik bozuklu�u (Child-Pugh S�n�f A ve B) olan hastalarda doz ayarlamas� gerekmemektedir. Bununla birlikte, hafif ve orta derecede hepatik bozuklu�u olan baz� bireylerde pirfenidonun plazma seviyesi artabilece�inden bu pop�lasyonda P�RFA® dikkatli kullan�lmal�d�r. Hastalar e�er pirfenidon ile birlikte bir CYPlA2 inhibit�r� de kullan�yorlarsa, toksisite belirtilerine kar�� s�kl�kla monit�rize edilmelidir. Pirfenidon, a��r hepatik bozuklu�u olan hastalarda veya son d�nem karaci�er hastal��� olan hastalarda �al���lmam��t�r ve bu hastalarda kullan�lmamal�d�r. Tedavi s�ras�nda karaci�er fonksiyonlar�n�n kontrol edilmesi ve art�� durumunda doz ayarlamas� yap�lmas� gerekebilir.
B�brek yetmezli�i:
Hafif veya orta dereceli renal bozuklu�u olan hastalarda doz ayarlamas� gerekmemektedir. Pirfenidon �iddetli renal yetmezli�i olan hastalarda (CrCl<30 ml/min) veya diyaliz gerektiren son d�nem renal bozuklu�u olan hastalarda kontrendikedir.
Pediyatrik pop�lasyon:
Pediyatrik pop�lasyonda �PF tedavisinde pirfenidon kullan�m�na ili�kin veri bulunmamaktad�r.
Geriyatrik pop�lasyon:
65 ya� ve �zeri hastalarda doz ayarlamas� gerekmemektedir.
4.3. Kontrendikasyonlar
Etkin madde
Pirfenidon ile anjiyo�dem hikayesi (bkz. b�l�m 4.4)
Fluvoksamin ile e� zamanl� kullan�mda (bkz. b�l�m 4.5)
�iddetli karaci�er yetmezli�i veya terminal d�nem karaci�er hastal��� olanlarda (bkz. b�l�m 4.2 ve 4.4)
�iddetli b�brek yetmezli�i (CrCl <30 ml/dak) veya diyalizi gerektiren son evre b�brek hastal��� (bkz. b�l�m 4.2 ve 5.2)
4.4. �zel kullan�m uyar�lar� ve �nlemleri
Pirfenidon kullan�m�n�n, fotogenotoksisite testlerinde ����a maruz kalma sonucunda anormal bir kromozom yap�s�na neden oldu�u g�sterilmi�tir; bu nedenle hastaya, ilac�n ����a maruz kalma �zerine ciltte kanser olu�umuna neden olabilme potansiyelinin a��klanmas� ve anlad���n�n s�zl� onay� al�nd�ktan sonra uygulanmas� �nemlidir.
Karaci�er fonksiyonu
Pirfenidon tedavisine ba�lamadan �nce karaci�er fonksiyon testleri (ALT, AST ve bilirubin) yap�lmal�d�r. Pirfenidon tedavisi boyunca ALT, AST ve bilirubin d�zeylerinde de�i�iklik meydana gelebilir. Bu nedenle tedavinin ilk 6 ayl�k d�neminde ayda 1 ve daha sonra her �� ayda bir karaci�er fonksiyon testleri yap�lmal�d�r (bkz. B�l�m 4.8).
Hepatik bozukluk
Orta derecede hepatik bozuklu�u olan ki�ilerde (�rn. Child-Pugh S�n�f B), pirfenidon maruziyeti % 60 artm��t�r. �nceden hafif ve orta derecede hepatik bozuklu�u bulunan hastalarda (�rn. Child-Pugh S�n�f A ve B) artan maruziyet ihtimaline kar�� pirfenidon dikkatli kullan�lmal�d�r. Hastalar toksisite belirtileri i�in, �zellikle pirfenidon ile birlikte CYP1A2 inhibit�r� bir ila� al�yorlarsa, s�kl�kla monit�rize edilmelidir. Pirfenidon, �iddetli hepatik bozuklu�u olan bireylerde �al���lmam��t�r ve �iddetli hepatik bozuklu�u olan hastalarda kullan�lmamal�d�r.
Fotosensitivite reaksiyonu ve ciltte k�zar�kl�k
P�RFA® tedavisi s�ras�nda direkt g�n �����na maruziyetten (g�ne� lambalar� dahil) ka��n�lmal�d�r veya en aza indirilmelidir. Hastalar, g�nl�k g�ne� koruyucu kullan�m�, g�ne�e maruziyeti engelleyecek k�yafetler giyme ve fotosensitiviteye neden olacak di�er t�bbi �r�nlerin kullan�m� konusunda bilgilendirilmelidir. Hastalar fotosensitivite veya ciltte
k�zar�kl�k semptomlar�n� hekimlerine bildirmeleri konusunda uyar�lmal�d�r. A��r fotosensitivite reaksiyonlar� yayg�n de�ildir. Hafiften a��ra fotosensitivite reaksiyonu veya ciltte k�zar�kl�k durumunda doz ayarlamas� veya ge�ici s�reyle tedaviye ara verme gerekebilir.
Anjiyo�dem
Nefes almada g��l�k veya h�r�lt�l� solunum ile birlikte g�r�lebilen y�zde, dudaklarda ve/veya dilde �i�me gibi anjiyo�dem raporlar� (baz�lar� ciddi) pazarlama sonras�nda pirfenidon kullan�m�yla birlikte g�r�lm��t�r. Bu sebeple, P�RFA® tedavisi uygulanan hastalarda anjiyo�dem belirtileri g�r�lmesi durumunda tedavi hemen kesilmelidir. Anjiyo�dem ge�iren hastalar�n tedavisi standart bak�m prosed�r�ne g�re ger�ekle�tirilmelidir. Pirfenidondan dolay� anjiyo�dem ge�mi�i olan hastalarda P�RFA® kullan�m� kontrendikedir (bkz. b�l�m 4.3).
Sersemlik
Pirfenidon kullanan hastalarda sersemlik raporlanm��t�r. Bu sebeple, dikkat ve koordinasyon gerektiren aktivitelerden �nce, hastalar bu t�bbi �r�n�n yol a�abilece�i sersemlik ile ilgili bilgilendirilmelidirler. Klinik �al��malarda, sersemlik ya�ayan hastalar�n �o�u tek bir olay tecr�be etmi� ve 22 g�nl�k ortalama devam s�resinde bir�ok olay da iyile�mi�tir. E�er sersemlik iyile�miyorsa veya a��rla��yorsa, doz ayarlamas� veya P�RFA® tedavisinin kesilmesi gereklidir.
Halsizlik
Pirfenidon alan hastalarda halsizlik raporlanm��t�r. Bu y�zden, dikkat ve koordinasyon gerektiren aktivitelerden �nce, hastalar bu t�bbi �r�n�n yol a�abilece�i halsizlik ile ilgili bilgilendirilmelidirler.
Kilo kayb�
Pirfenidon alan hastalarda kilo kayb� rapor edilmi�tir. Hekimler hastan�n a��rl���n� takip etmeli ve kilo kayb� klinik anlaml�l�k g�sterdi�inde hastan�n uygun kalori al�m�n� sa�lamal�d�r.
P�RFA®, laktoz monohidrat (inek s�t�) i�ermektedir. Nadir kal�t�msal galaktoz intolerans�, Lapp laktoz yetmezli�i ya da glikoz-galaktoz malabsorpsiyon problemi olan hastalar�n bu ilac� kullanmamalar� gerekir.
4.5. Di�er t�bbi �r�nler ile etkile�imler ve di�er etkile�im �ekilleri
Pirfenidonun yakla��k % 70-80'i, CYP2C9, 2C19, 2D6 ve 2E1'i i�eren di�er CYP izoenzimlerinin min�r katk�lar�yla, CYP1A2 taraf�ndan metabolize edilmektedir.
Greyfurt suyu t�ketimi CYP1A2'nin inhibisyonu ile ili�kilidir bu nedenle pirfenidon ile tedavi s�ras�nda greyfurt suyu t�ketiminden ka��n�lmal�d�r.
Fluvoksamin ve CYP1A2 inhibit�rleri
Faz 1 �al��mas�nda, pirfenidon ve fluvoksaminin [di�er CYP izoenzimleri (CYP2C9, 2C19 ve 2D6) �zerinde inhibit�r etkilere sahip CYP1A2'nin g��l� bir inhibit�r�] birlikte kullan�lmas�, sigara i�meyen hastalar�n pirfenidon maruziyetinde 4-kat art��la sonu�lanm��t�r.
E� zamanl� fluvoksamin kullanan hastalarda pirfenidon kontrendikedir (bkz. b�l�m 4.3). Pirfenidon tedavisine ba�lamadan �nce fluvoksamin tedavisi kesilmeli ve pirfenidon tedavisi s�ras�nda pirfenidonun azalm�� klerensi nedeniyle fluvoksamin kullan�m�ndan ka��n�lmal�d�r. Pirfenidon tedavisi s�ras�nda hem CYP1A2'nin hem de pirfenidon metabolizmas�nda yer alan bir veya daha fazla di�er CYP izoenzimi (�rn. CYP2C9, 2C19 ve 2D6)'nin inhibit�r� olan di�er tedavilerden ka��n�lmal�d�r.
�n vitro ve in vivo ekstrapolasyonlar, CYP1A2'nin g��l� ve se�ici inhibit�rlerinin (�rne�in, enoxacin), pirfenidona maruziyeti yakla��k 2 ila 4 kat art�rma potansiyeline sahip oldu�unu g�stermektedir. E�er pirfenidonun g��l� ve se�ici CYP1A2 inhibit�r� ile birlikte kullan�lmas� �nlenemezse, pirfenidonun dozu g�nde 800 mg'a azalt�lmal�d�r (g�nde d�rt kez 200 mg). Pirfenidon tedavisiyle ili�kili advers reaksiyonlar a��s�ndan hastalar yak�ndan izlenmelidir. Gerekirse pirfenidon tedavisi durdurulmal�d�r (bkz. b�l�m 4.2 ve 4.4).
Pirfenidon ve 750 mg siprofloksasinin birlikte uygulanmas� (orta derecede CYP1A2 inhibit�r�) pirfenidona maruz kalmay� % 81 oran�nda art�r�r. G�nde iki kez 750 mg dozunda siprofloksasin al�m� gerekli ise, pirfenidonun dozu g�nde 1600 mg'a d���r�lmelidir (g�nde d�rt kez 400 mg). Pirfenidon, siprofloksasin g�nde bir veya iki kez 250 mg veya 500 mg'l�k bir dozda kullan�ld���nda dikkatli kullan�lmal�d�r.
Pirfenidon, di�er orta derecede CYP1A2 inhibit�rleri (�rn. amiodaron, propafenon) ile tedavi edilen hastalarda dikkatli kullan�lmal�d�r.
CYP2C9 (�rn. amiodaron, flukonazol), 2C19 (�rn. kloramfenikol) ve 2D6 (�rn. fluoksetin paroksetin) gibi pirfenidonun metabolizmas�nda yer alan bir veya daha fazla CYP izoenziminin g��l� inhibit�rleriyle CYP1A2 inhibit�rleri e� zamanl� olarak kullan�ld���nda �zel dikkat g�sterilmelidir.
CYP1A2 ind�kleyicileri
Orta derecede CYP1A2 ind�kleyicileri (�rne�in omeprazol), e�zamanl� kullan�ld���nda teorik olarak pirfenidon plazma seviyelerinin d��mesine neden olabilir.
Hem CYP1A2 ind�kleyicilerinin hem de pirfenidonun metabolizmas�nda rol oynayan di�er CYP izoenzimlerinin g��l� ind�kleyicileri olarak etki g�steren t�bbi �r�nlerin birlikte uygulanmas�, pirfenidon plazma seviyelerinin �nemli �l��de d��mesine neden olabilir.
M�mk�nse bu t�bbi �r�nlerin kullan�m�ndan ka��n�lmal�d�r.
�zel pop�lasyonlara ili�kin ek bilgiler Pediyatrik pop�lasyon
Pirfenidonun, d���k do�um a��rl���na sahip bebeklerde, yeni do�an bebeklerde, emzirilen yeni do�anlarda, bebeklerde veya �ocuklarda g�venlili�i belirlenmemi�tir.
Geriyatrik pop�lasyon
Ya�l� hastalarda fizyolojik fonksiyon genellikle gerilemi�tir; bu nedenle pirfenidon dikkatle uygulanmal�d�r.
4.6. Gebelik ve laktasyon
Gebelik kategorisi: C
�ocuk do�urma potansiyeli bulunan kad�nlar/Do�um kontrol� (Kontrasepsiyon) Pirfenidonun �ocuk do�urma potansiyeli bulunan kad�nlar ve do�um kontrol� �zerine olan etkisi bilinmemektedir. �ocuk do�urma potansiyeline sahip kad�n hastalara tedavi s�ras�nda etkili bir do�um kontrol y�ntemi kullanmalar� �nerilmelidir.
Gebelik d�nemi
Hamilelerde pirfenidon kullan�m�na dair bir veri bulunmamaktad�r.
Hayvanlarda pirfenidonun ve/veya metabolitlerinin plasentadan transferi, pirfenidonun ve/veya metabolitlerinin amniyotik s�v�da birikmesi potansiyeli ile meydana gelir.
Y�ksek dozlarda (≥1000 mg/kg/g�n) s��anlarda gebelik s�resinde uzama ve f�tal ya�am �ans�nda azalma g�r�lm��t�r.
Tedbirli olmak ad�na, hamilelik s�ras�nda pirfenidon kullan�lmas�ndan ka��n�lmal�d�r.
Laktasyon d�nemi
Pirfenidonun veya metabolitlerinin insan s�t�ne ge�ip ge�medi�i bilinmemektedir. Hayvanlardan elde edilen farmakokinetik veriler, pirfenidonun ve/veya metabolitlerinin s�te ge�mesi ve beraberinde s�tte birikimi potansiyelini g�stermektedir. Risk g�z ard� edilemez.
Emzirmenin �ocu�a faydas� ve pirfenidon terapisinin anneye faydas� dikkate al�narak, emzirmenin kesilmesi veya pirfenidon terapisinin kesilmesi y�n�nde bir karar al�nmal�d�r.
�reme yetene�i/Fertilite
Klinik �ncesi �al��malarda, fertilite �zerinde advers etkiler g�zlemlenmemi�tir.
4.7. Ara� ve makine kullan�m� �zerindeki etkiler
P�RFA® sersemlik ve halsizli�e sebep olabilir, bu da ara� ve makine kullan�m�n� etkileyebilir.
4.8. �stenmeyen etkiler
Pirfenidonun g�venlili�i 1650 g�n�ll� ve hasta ile klinik �al��ma ile de�erlendirilmi�tir. I70'ten fazla hasta be� y�ldan fazla s�re ve bir k�sm� da on y�la kadar, a��k �al��malarda ara�t�r�lm��t�r.
Pirfenidon ile 2403 mg/g�n dozda, plasebo kar��la�t�rmal� klinik �al��ma esnas�nda en yayg�n raporlanan advers reaksiyonlar s�ras�yla a�a��daki gibidir:
Advers Reaksiyon | Pirfenidon 2403 mg/g�n | Plasebo |
Bulant� | % 32,4 | % 12,2 |
Ciltte k�zar�kl�k | % 26,2 | % 7,7 |
Diyare | % 18,8 | % 14,4 |
Halsizlik | % 18,5 | % 10,4 |
Dispepsi | % 16,1 | % 5,0 |
Anoreksiya | % 11,4 | % 3,5 |
Ba� a�r�s� | % 10,1 | % 7,7 |
Fotosensitivite reaksiyonu | % 9,3 | % 1,l |
Ciddi advers reaksiyonlar 2403 mg/g�n pirfenidon ile tedavi edilen hastalar ve plasebo alanlar aras�nda benzer s�kl�klarda kaydedilmi�tir.
�� pivotal Faz 3 �al��mas�nda �nerilen doz olan 2403 mg/g�n pirfenidon kullanan 623 hastada ≥% 2 s�kl���nda raporlanan advers reaksiyonlar Tablo l'de g�sterilmi�tir. Pazarlama sonras� �al��malardan elde edilen advers reaksiyonlar da Tablo l'de listelenmi�tir. Advers reaksiyonlar Sistem Organ S�n�fi ve s�kl�k grupland�rmas�na g�re listelenmi�tir [�ok yayg�n (≥1/10), yayg�n (≥l/100 ila <1/10), yayg�n olmayan (≥l/l.000 ila <1/100), seyrek ≥1/l0.000 ila
<1/1.000), �ok seyrek (≤ 1/10.000), bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor)] advers reaksiyonlar azalan ciddiyetlerine g�re s�ralanm��t�r.
Tablo 1 | |
Enfeksiyonlar ve enfestasyonlar | |
Yayg�n: | �st solunum yollar� enfeksiyonlar�, idrar yolu enfeksiyonlar� |
Kan ve lenf sistemi hastal�klar� | |
Seyrek: | Agran�lositoz |
�mm�n sistem hastal�klar� | |
Yayg�n olmayan: | Anjiyo�dem |
Metabolizma ve beslenme hastal�klar� | |
�ok yayg�n: | Anoreksiya |
Yayg�n: | Kilo kayb�, i�tah azalmas� |
Psikiyatrik hastal�klar | |
Yayg�n: | �nsomniya |
Sinir sistemi hastal�klar� | |
�ok yayg�n: | Ba� a�r�s� |
Yayg�n: | Sersemlik, somnolans, disguzi, letarji |
Vask�ler hastal�klar | |
Yayg�n: | S�cak basmas� |
Solunum, g���s bozukluklar� ve mediastinal hastal�klar | |
Yayg�n: | Dispne, �ks�r�k, balgaml� �ks�r�k |
Gastrointestinal hastal�klar | |
�ok yayg�n: | Dispepsi, bulant�, diyare |
Yayg�n: | Gastro�zofagal refl� hastal���, kusma, abdominal �i�kinlik, abdominal rahats�zl�k, abdominal a�r�, �st abdominal a�r�, mide rahats�zl���, gastrit, konstipasyon, midede gaz |
Hepato-biliyer hastal�klar� | |
Yayg�n: | ALT/AST d�zeylerinde de�i�iklik, gama glutamil trasferaz art��� |
Seyrek: | ALT ve ASTd�zeylerinde de�i�iklik ile birlikte total serum bilirubin art��� |
Deri ve deri alt� doku hastal�klar� | |
�ok yayg�n: | Fotosensitivite reaksiyonu, d�k�nt� |
Yayg�n: | Ka��nt�, eritema, kuru cilt, eritematoz d�k�nt�, mak�ler d�k�nt�, ka��nt�l� d�k�nt� |
Kas-iskelet bozukluklar�, ba� doku ve kemik hastal�klar� | |
Yayg�n: | Miyalji, artralji |
Genel bozukluklar ve uygulama b�lgesine ili�kin hastal�klar | |
�ok yayg�n: | Halsizlik |
Yayg�n: | Asteni, kardiyak olmayan g���s a�r�s� |
Yaralanma ve zehirlenme | |
Yayg�n: | G�ne� yan��� |
1 pazarlama sonras� anketlerden elde edilmi�tir.
��pheli advers reaksiyonlar�n raporlanmas�
Ruhsatland�rma sonras� ��pheli ila� advers reaksiyonlar�n�n raporlanmas� b�y�k �nem ta��maktad�r. Raporlama yap�lmas�, ilac�n yarar/risk dengesinin s�rekli olarak izlenmesine olanak sa�lar. Sa�l�k mesle�i mensuplar�n�n herhangi bir ��pheli advers reaksiyonu T�rkiye Farmakovijilans Merkezi (T�FAM)'ne bildirmeleri gerekmektedir (www.titck.gov.tr; e- posta: tufam@titck.gov.tr; tel: 0 800 314 00 08; faks: 0 312 218 35 99)
4.9. Doz a��m� ve tedavisi
Doz a��m� konusunda yetersiz klinik bilgi mevcuttur. Pirfenidonun g�nde 4806 mg'a kadar olan �oklu dozlar�, g�nde �� defa alt� adet 267 mg'l�k kaps�l olarak sa�l�kl� eri�kin g�n�ll�lere 12-g�nl�k doz art�rma periyodundan sonra uygulanm��t�r. Advers reaksiyonlar hafif, ge�ici ve pirfenidon i�in en s�k rapor edilen advers reaksiyonlar ile tutarl�d�r.
��phelenilen bir doz a��m� halinde, ya�amsal i�aretlerin izlenmesi ve hastan�n klinik durumunun yak�ndan izlenmesi dahil destekleyici t�bbi bak�m sa�lanmal�d�r.
5. FARMAKOLOJ�K �ZELL�KLER
5.1. Farmakodinamik �zellikler
Farmakoterap�tik grup: Di�er imm�nosupresanlar ATC kodu: L04AX05
Pirfenidonun etki mekanizmas� tam olarak belirlenmemi�tir. Bununla birlikte, �e�itli in vitro sistemlerde ve pulmoner fibr�z hayvan modellerinde (bleomisin- ve transplant-ind�klenmi�
fibr�z) veriler pirfenidonun hem antifibrotik hem de antiinflamatuvar �zellikler ortaya koydu�unu g�sterir.
�diyopatik pulmoner fibrozis (�PF), t�m�r nekroz fakt�r�-alfa (TNF-α) ve interl�kin-1-beta (IL-1β) dahil proenflamatuvar sitokinlerin sentezi ve sal�m� taraf�ndan etkilenen bir kronik fibrotik ve enflamatuvar pulmoner hastal�kt�r ve pirfenidonun �e�itli uyaranlara yan�t olarak inflamatuvar h�crelerin birikmesini azaltt��� g�sterilmi�tir.
Pirfenidon, fibroblast proliferasyonunu fibrozis ile ili�kili proteinlerin ve sitokinlerin �retimini ve d�n��t�r�c� b�y�me fakt�r�-beta (TGF-β) ve trombosit kaynakl� b�y�me fakt�r� (PDGF) gibi sitokin b�y�me fakt�rlerine yan�t olarak h�cre-d��� matrisin biyosentezindeki ve birikmesindeki art��� azalt�r.
Sa�kal�m
Pirfenidonun plaseboya k�yasla sa�kal�m�, �al��ma 1, 2 ve 3'te primer son noktay� desteklemek amac�yla ke�ifsel analiz �eklinde hesaplanm��t�r. T�m nedenlere ba�l� mortalite, �al��ma s�resince ve mevcut takip periyodunda, �l�m�n sebebine ve hastan�n tedaviye devam edip etmedi�ine bakmaks�z�n de�erlendirilmi�tir. T�m nedenlere ba�l� mortalite, istatistiksel olarak anlaml� bir farkl�l�k g�stermemi�tir (bkz. Grafik 1).
Grafik 1. Vital Durumda T�m Nedenlere Ba�l� Mortalite Kaplan-Meier Tahminleri- �al��ma Sonu: �al��ma 1, 2 ve 3

Klinik etkililik
Pirfenidonun klinik etkilili�i �PF'li hastalarda d�rt Faz 3, �ok merkezli, randomize, �ift-k�r, placebo-kontroll� �al���lm��t�r. Faz 3 �al��malar�n�n ��� (PIPF-004, PIPF-006 ve PIPF-016) �ok-uluslu, biri (SP-3) Japonya'da y�r�t�lm��t�r.
PIPF-004 ve PIPF-006, pirfenidon 2403 mg/g�n tedavisi ile plasebonun kar��la�t�r�lmas�d�r. �al��malar�n tasar�m�, PIPF-004'te ara doz grubunu (1197 mg/g�n) i�eren birka� istisna
d���nda neredeyse ayn�d�r. �ki �al��mada da, tedavi en az 72 hafta boyunca g�nde �� defa uygulanm��t�r. �ki �al��mada da primer var�� noktas�, ba�lang��tan 72. haftaya kadar, y�zde beklenen Zorlu Vital Kapasite (Forced Vital Capacity, FVC)'nin de�i�imidir.
PIPF-004 �al��mas�nda, pirfenidon alan hastalarda (N=174), placebo alan hastalara k�yasla (N=174, p=0,001) ba�lang��tan tedavinin 72. haftas�na kadar, beklenen FVC d����� y�zdesi belirgin bir �ekilde azalm��t�r. Pirfenidon tedavisi ba�lang��tan 24. (p=0,014), 36. (p<0,001),
48. (p<0,001) ve 60. (p<0,001) haftalara kadar da, beklenen FVC d����� y�zdesini belirgin bir �ekilde azaltm��t�r. 72. haftada, beklenen FVC y�zdesinde ba�lang��tan d���� ≥% 10 (�PF mortalite riskinin indikat�r e�i�i) Pirfenidon alan hastalarda % 20, plasebo alan hastalarda % 35 olarak g�zlenmi�tir (Tablo 2).
Tablo 2 PIPF-004 �al��mas�nda ba�lang��tan 72. haftaya beklenen FVC y�zdesi de�i�iminin kategorik de�erlendirmesi | ||
| Pirfenidon 2403 mg/g�n (N=174) | Plasebo (N=174) |
≥% 10 azalma veya �l�m veya akci�er nakli | 35 (% 20) | 60 (% 35) |
% 10'dan daha az azalma | 97 (% 56) | 90 (% 52) |
Azalma yok (FVC de�i�imi > % 0) | 42 (% 24) | 24 (% 14) |
Pirfenidon alan hastalarda placebo alan hastalara k�yasla rank ANCOVA ile �nceden belirlenmi� alt� dakika y�r�y�� testi (6MWT) s�ras�nda ba�lang��tan 72. haftaya kadar bir fark olmasa da, rastgele bir analizde Pirfenidon alan hastalar�n % 37'sinde, PIPF-004'teki plasebo alan hastalar�n % 47'sine k�yasla 6MWT'de ≥50 m azalma g�zlenmi�tir.
PIPF-006 �al��mas�nda, Pirfenidon tedavisi (N=171) ba�lang��tan 72. haftaya kadar, plasebo ile kar��la�t�r�ld���nda (N=173; p=0,501) y�zde beklenen FVC d�����n� azaltmam��t�r. Bununla beraber, pirfenidon tedavisi ba�lang��tan 24. (p<0,001), 36. (p=0,011) ve 48. (p=0,005) haftaya kadar y�zde beklenen FVC d�����n� azaltm��t�r. 72. Haftada, FVC'de ≥%
10 d���� pirfenidon alan hastalar�n % 23'�nde ve plasebo alan hastalar�n % 27'sinde g�r�lm��t�r (Tablo 3).
Tablo 3 PIPF-006 �al��mas�nda ba�lang��tan 72. haftaya de�i�iminin kategorik de�erlendirmesi | y�zde beklenen FVC | |
| Pirfenidon 2403 mg/g�n (N=171) | Plasebo (N=173) |
≥% 10 azalma veya �l�m veya akci�er nakli | 39 (% 23) | 46 (% 27) |
% 10'dan daha az azalma | 88 (% 52) | 89 (% 51) |
Azalma yok (FVC de�i�imi > % 0) | 44 (% 26) | 38 (% 22) |
Ba�lang��tan 72. haftaya kadar 6MWT uzakl���ndaki d���� PIPF-006 (p<0,001, rank ANCOVA) �al��mas�ndaki plasebo ile k�yasland���nda belirgin bir �ekilde azalm��t�r. Ek olarak, bir ad hoc analizde, pirfenidon alan hastalar�n %33'�, 6MWT mesafesinde ≥ 50 m'lik bir d���� g�sterdi, bu da PIPF-006'da plasebo alan hastalar�n % 47'sine kar��l�k gelmektedir.
PIPF-004 ve PIPF-006'dan elde edilen havuzlanm�� hayatta kalma analizinde, �l�m oran� pirfenidon 2403 mg/g�n grubunda % 7,8 iken, plasebo alan grupta % 9,8 olarak hesaplanm��t�r (HR 0,77 [%95 CI, 0,47-1,28]).
PIPF-016, 2403 mg/g�n Pirfenidonun plaseboya kar�� kar��la�t�r�lmal� �al��mas�d�r. Tedavi g�nde �� defa 52 hafta boyunca uygulanm��t�r. Primer var�� noktas� ba�lang��tan 52. haftaya kadar y�zde beklenen FVC de�i�imidir. 555 hastan�n tamam�nda, ortalama ba�lang�� y�zde beklenen FVC ve % DLco s�ras�yla % 68 (aral�k % 48-91) ve % 42 (aral�k % 27-170)'dir. Hastalar�n y�zde ikisinde y�zde beklenen FVC % 50'nin alt�ndad�r ve hastalar�n % 21'inde y�zde beklenen DLco ba�lang��ta % 35'in alt�ndad�r.
PIPF-016 �al��mas�nda, ba�lang��tan tedavinin 52. haftas�na kadar y�zde beklenen FVC d�����, Pirfenidon alan hastalarda (N=278), plasebo alan hastalarla (N=277; p<0,000001, rank ANCOVA) k�yasland���nda, belirgin bir �ekilde azalm��t�r. Pirfenidon ile tedavi ba�lang��tan tedavinin 13. (p<0,000001), 26. (p<0,000001) ve 39. (p=0,000002) haftas�na kadar y�zde beklenen FVC d����� belirgin bir �ekilde azalm��t�r. 52. haftada, FVC'de ≥% 10 d���� Pirfenidon alan hastalar�n % 17'�nde ve plasebo alan hastalar�n % 32'sinde g�r�lm��t�r (Tablo 4).
Tablo 4 PIPF-016 �al��mas�nda ba�lang��tan 52. haftaya y�zde beklenen FVC de�i�iminin kategorik de�erlendirmesi | ||
| Pirfenidon 2403 mg/g�n (N=278) | Plasebo (N=277) |
≥% 10 azalma veya �l�m | 46 (% 17) | 88 (% 32) |
% 10'dan daha az azalma | 169 (% 61) | 162 (% 58) |
Azalma yok (FVC de�i�imi > % 0) | 63 (% 23) | 27 (% 10) |
Ba�lang��tan 52. haftaya kadar 6MWT uzakl���ndaki d���� PIPF-016 (p<0,036, rank ANCOVA) �al��mas�ndaki plasebo ile k�yasland���nda belirgin bir �ekilde azalm��t�r; Pirfenidon alan hastalar�n % 26's�, plasebo alan hastalar�n % 36's� ile kar��la�t�r�ld���nda, 6MWT uzakl���nda ≥50 m d���� g�stermi�tir.
PIPF-016, PIPF-004 ve PIPF-006 �al��malar�n�n 12. ayda �nceden belirlenmi� toplam analizinde, t�m sebep-olunmu� mortalite, Pirfenidon 2403 mg/g�n grubunda (% 3,5, 623 hastan�n 22'si) plasebo alan hastalarla (% 6,7, 624 hastan�n 42'si) k�yasland���nda mortalite belirgin bir �ekilde azd�r, sonu�lanan % 48'inde ilk 12 ayda t�m sebep olunan mortalite riskinde azalma g�zlenmi�tir (HR 0,52 [% 95 Cl, 0,31-0,87], p=0,0107, log-rank test).
Japon hastalarda yap�lan �al��mada (SP3), pirfenidon 1800 mg/g�n (ABD'de ve PIPF- 004/006'n�n Avrupa pop�lasyonunda, 2403 mg/g�n ile a��rl�k-normalize edilmi� olarak kar��la�t�r�labilir) plasebo ile kar��la�t�r�lm��t�r (s�ras�yla N = 110, N = 109). Pirfenidon ile tedavi vital kapasitede (VC) ortalama d����� 52. haftada (primer var�� noktas�) plasebo ile k�yasland���nda belirgin bir �ekilde azalm��t�r (-0,09±0,02 I kar�� -0,16±0,02 I s�ras�yla, p=0,042).
Pediyatrik pop�lasyon
Avrupa �la� Ajans�, �PF'li pediyatrik pop�lasyonun t�m alt k�melerinde pirfenidon ile �al��malar�n sonu�lar�n�n yay�nlanmas� y�k�ml�l���nden vazge�mi�tir.
5.2. Farmakokinetik �zellikler
Genel �zelliklerEmilim:
Pirfenidon kaps�llerin g�da ile verilmesi, C'ta b�y�k bir azalma (% 50 oran�nda) ve EAA'da a�l�k durumuna k�yasla daha k���k bir etki ile sonu�lan�r. Tokluk durumunda sa�l�kl� ya�l� yeti�kin g�n�ll�lere (50-66 ya�) tek bir doz 801 mg oral yoldan uyguland�ktan sonra, pirfenidon emilim h�z� yava�larken tokluk durumdaki EAA de�eri a�l�k durumundakinin yakla��k % 80-85'i olarak hesaplanm��t�r. A�l�k durumunda 801 mg tabletin �� 267 mg'l�k kaps�l ile kar��la�t�r�ld��� �al��mada biyoe�de�erlik g�sterilmi�tir. Tokluk durumda, 801 mg tablet, kaps�llere k�yasla EAA �l��mlerine dayanan biyoe�de�erlik kriterlerini kar��larken, Ci�in % 90 g�ven aral��� (% 108,26 - % 125,60), standart biyoe�de�erlik s�n�r�n�n) �st s�n�r�n� biraz a�m��t�r (% 90 CI: % 80,00 - % 125,00). G�dalar�n oral pirfenidon EAA �zerindeki etkisi, tablet ve kaps�l form�lasyonlar� aras�nda tutarl�yd�.
A�l�k durumuna k�yasla, her iki form�lasyonun da g�da ile uygulanmas� pirfenidon Cde�erini azalt�rken, pirfenidon tabletin Cde�eri pirfenidon kaps�llere g�re biraz daha az (% 50) azaltm��t�r (% 40'a kar��l�k % 50).
A� karna ila� alan gruba k�yasla, tok karna ila� alan bireylerde, d���k yan etki insidans� (bulant� ve sersemlik) g�zlenmi�tir. Bu nedenle, pirfenidon'un bulant� ve sersemlik insidans�n� azaltmak i�in g�da ile birlikte uygulanmas� tavsiye edilir.
�nsanlarda pirfenidonun mutlak biyoyararl�l��� belirlenmemi�tir.
Plazma Konsantrasyonlar�
A� karn�na al�nan tek doz
Alt� sa�l�kl� erkek yeti�kine, a� karn�na tek bir oral uygulama ile verilen 200 mg, 400 mg ve 600 mg pirfenidonun plazma konsantrasyonlar� ve farmakokinetik parametreleri �ekil 1'de ve Tablo 1'de g�sterilmektedir.
Hem Chem de EAA, doz ile yakla��k olarak kar��la�t�r�ld���nda artm��t�r.
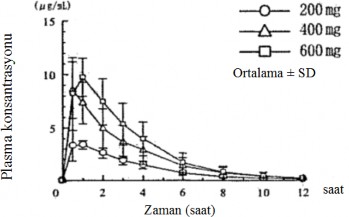
Doz miktar� (mg) | C(μg/ml) | T(saat) | EAA (μg·saat/ml) | T(saat) |
200 | 3,88 ± 0,82 | 0,75 ± 0,27 | 13,97 ± 2,71 | 2,10 ± 0,45 |
400 | 9,24 ± 1,74 | 0,58 ± 0,20 | 29,10 ± 11,77 | 1,96 ± 0,55 |
600 | 10,57 ± 1,78 | 0,83 ± 0,26 | 37,03 ± 11,97 | 1,76 ± 0,40 |
�ekil 1: A� karn�na al�nan tek bir dozun ard�ndan plasma konsantrasyonlar� Tablo 1: Farmakokinetik parametreler (n=6)
(�l��m y�ntemi: HPLC) (Ortalama ± SD)
5.3. Klinik �ncesi g�venlilik verileri
G�venlik farmakolojisi, tekrarlanan doz toksisitesi, genotoksisite ve karsinojenik potansiyel ile ilgili konvansiyonel �al��malara dayal� klinik d��� veriler, insanlar i�in �zel bir tehlike olmad���n� ortaya koymaktad�r.
Tekrarl� doz toksisite �al��malar�nda, fare, s��an ve k�peklerde karaci�er a��rl�k art��� g�zlenmi�tir; genellikle hepatik sentrilobular hipertropi ile birlikte g�r�l�r. Tedavinin kesilmesinden sonra geri d�n�� g�zlenmi�tir. S��anlarda ve farelerde y�r�t�len karsinojenite �al��malar�nda karaci�er t�m�r� insidans�nda art�� g�zlenmi�tir. Bu hepatik bulgular hepatik mikrozomal enzimlerin ind�ksiyonu ile ba�lant�l�d�r, bu etki Pirfenidon alan hastalarda g�zlenmemi�tir. Bu bulgular�n insanlar ile ili�ki oldu�u d���n�lmemektedir.
�nsan 2403 mg/g�n dozunun 37 kat� olan 1500 mg/kg/g�n doz uygulanan di�i s��anlarda uterus t�m�rlerinde istatistiksel olarak belirgin art�� g�zlenmi�tir. Hastal�k geli�me mekanizmas� ile ilgili �al��malar�n sonu�lar�na g�re, uterus t�m�rlerinin g�r�lmesi muhtemelen s��anlardaki t�re spesifik endokrin metabolizmas�n� i�eren kronik dopamin- arac�l� seks hormon dengesizli�inden dolay�d�r, bu da insanlarda bulunmamaktad�r.
Reprod�ktif toksikoloji �al��malar�nda, di�i veya erkek s��an fertilitesinde veya yavrular�n postnatal geli�imi �zerinde advers etki g�r�lmemi�tir ve s��anlarda (1000 mg/kg/g�n) veya tav�anlarda (300 mg/kg/g�n) teratojenite g�r�lmemi�tir. Hayvanlarda pirfenidonun ve/veya metabolitlerinin plasental transferi, pirfenidonun ve/veya metabolitlerinin amniyotik s�v�da birikmesi ile ortaya ��kar. Y�ksek dozlarda (≥450 mg/kg/g�n) s��anlarda �strus d�ng�s�nde uzama ve d�zensiz d�ng�lerin insidans�nda y�kselme g�zlenmi�tir. Y�ksek dozlarda (≥1000 mg/kg/g�n) s��anlarda hamilelik s�resinde uzama ve f�tal ya�am �ans�nda azalma g�zlenmi�tir.
Emziren s��anlardaki �al��malar, pirfenidonun ve/veya metabolitlerinin s�te at�l�m�n�n, pirfenidonun ve/veya metabolitlerinin s�tte birikmesi potansiyelleri oldu�unu g�stermi�tir.
Pirfenidon standart testler grubunda mutajenik veya genotoksik aktivite g�stermemi�tir ve UV maruziyet alt�nda test edildi�inde mutajenik de�ildir. UV maruziyet alt�nda test edildi�inde Pirfenidon, �in hamster� akci�er h�cresinde fotoklastojenik miktar tayininde pozitif ��km��t�r.
Oral yoldan pirfenidon verilen kobaylarda, UVA/UVB �����na maruz b�rak�ld�klar�nda fototoksisite ve iritasyon geli�ti�i kaydedilmi�tir. G�ne� koruyucu uygulamas�yla fototoksisite lezyonlar�n�n �iddeti minimize edilmi�tir.
6. FARMAS�T�K �ZELL�KLER
6.1. Yard�mc� maddelerin listesi
Laktoz monohidrat (inek s�t�) Hidroksipropil sel�lozKarmelloz kalsiyum Magnezyum stearat
Film kaplama materyal (Materyal i�eri�i: Hidroksipropil sel�loz, Titanyum dioksit (E171), Trietil sitrat, Talk, Magnezyum stearat, K�rm�z� demir oksit (E172), Sar� demir oksit (E172), Siyah demir oksit (E172))
6.2. Ge�imsizlikler
Bilinen herhangi bir ge�imsizli�i bulunmamaktad�r.
6.3. Raf �mr�
24 ay
6.4. Saklamaya y�nelik �zel tedbirler
25°C alt�ndaki oda s�cakl���nda saklay�n�z.
6.5. Ambalaj�n niteli�i ve i�eri�i
Kutuda, �effaf PVC/PE/PVDC – Alu blister ambalajlarda
252 film tabletlik blister ambalajlarda takdim edilmektedir.
6.6. Be�eri t�bbi �r�nden arta kalan maddelerin imhas� ve di�er �zel �nlemler
Kullan�lmam�� olan �r�nler ya da at�k materyaller “T�bbi At�klar�n Kontrol� Y�netmeli�i” ve “Ambalaj ve Ambalaj At�klar�n�n Kontrol� Y�netmelikleri”ne uygun olarak imha edilmelidir.
 Ruh ve Ak�l Sa�l���m�z� Geli�tirmek
�yi ak�l ve ruh sa�l��� sahip olmaktan ziyade, yapt���n�z �eylerdir. Ak�l ve
ruhsal olarak sa�l�kl� olmak i�in kendinize de�er vermeli ve kendinizi kabul
etmelisiniz.
Ruh ve Ak�l Sa�l���m�z� Geli�tirmek
�yi ak�l ve ruh sa�l��� sahip olmaktan ziyade, yapt���n�z �eylerdir. Ak�l ve
ruhsal olarak sa�l�kl� olmak i�in kendinize de�er vermeli ve kendinizi kabul
etmelisiniz. |
 �nme
�nme, beynin hasar g�rmesinin sonucudur. Bu hasar, beynin bir k�sm�ndaki ya bir kanama
ya da akut kan eksikli�i nedeniyle o k�sm�n ge�ici ya da kal�c� olarak i�levini yapamamas�na
yol a�ar.
�nme
�nme, beynin hasar g�rmesinin sonucudur. Bu hasar, beynin bir k�sm�ndaki ya bir kanama
ya da akut kan eksikli�i nedeniyle o k�sm�n ge�ici ya da kal�c� olarak i�levini yapamamas�na
yol a�ar. |
�LA� E�DE�ERLER�
| E�de�er �la� Ad� | Barkodu | �la� Fiyat� |
|---|---|---|
| MONTENIDON | 8680835090128 | 27,826.62TL |
| PIRFA | 8699543091853 | 29,732.97TL |
| PIRFECT | 8699540090972 | 32,366.14TL |
| Di�er E�de�er �la�lar |
 |
Grip, So�uk Alg�nl��� ve �ks�r�k Grip ve so�uk alg�nl��� (nezle) semptomlar� aras�ndaki fark� bilmek �nemlidir. So�uk alg�nl��� gripten daha hafif belirtiler g�steren bir solunum yolu hastal���d�r. |
 |
�izofrenlik �izofrenli�in psikiatrik te�hisi hakk�nda �ok fazla anla�mazl�k vard�r. Bu sayfadaki bilgiler, �izofrenli�in te�hisi, nedenleri ve tedavisi hakk�ndaki fakl� teoriler hakk�nda bilgi verecektir. |
 |
Do�um Sonras� Depresyonu Do�um sonras� depresyonu, do�umdan sonra her on kad�ndan biri taraf�ndan tecr�be edilen stresli bir durumdur. |
�LA� GENEL B�LG�LER�
Ali Raif �la� San. A.�.
| Geri �deme Kodu | A17171 |
| Sat�� Fiyat� | 29732.97 TL [ 3 Aug 2026 ] |
| �nceki Sat�� Fiyat� | 29732.97 TL [ 27 Jul 2026 ] |
| Original / Jenerik | Jenerik �la� |
| Re�ete Durumu | Normal Re�eteli bir ila�d�r. |
| Barkodu | 8699543091853 |
| Etkin Madde | Pirfenidon |
| ATC Kodu | L04AX05 |
| Birim Miktar | 200 |
| Birim Cinsi | MG |
| Ambalaj Miktar� | 252 |
| Antineoplastik ve �mm�nomod�lat�r Ajanlar > �mm�nsupresif Ajanlar > Pirfenidone |
| Yerli ve Be�eri bir ila�d�r. |