NEXAVAR 200 mg 112 film tablet Kısa Ürün Bilgisi
{ Sorafenib }
1. BEŞERİ TIBBİ ÜRÜNÜN ADI
NEXAVAR® 200 mg film kaplı tablet2. KALİTATİF VE KANTİTATİF BİLEŞİM
Sorafenib 200 mg (274 mg sorafenib tosilat olarak)
Yardımcı maddeler için 6.1.’e bakınız.
Yardımcı maddeler için 6.1.’e bakınız.
3. FARMASÖTİK FORMU
Film kaplı tablet.
Bir yüzünde Bayer logosu, diğer yüzünde "200" ifadesi bulunan, 10 mm çapında ve 350 mg ağırlığında, yuvarlak, bikonveks, kırmızı tabletler.
4.1. Terapötik endikasyonlar
NEXAVAR®;
- Metastatik renal hücreli karsinoma (RCC) tedavisinde interferon alfa ve/veya interlökin-2 ile yanıt alınamadığı veya bu tip tedavilerin yan etki nedeniyle uygun olmadığı durumlarda kullanılması için endikedir.
4.2. Pozoloji ve uygulama şekli
Erişkinler:
Pozoloji
Önerilen NEXAVAR® dozu günde toplam 800 mg’dır.
Uygulama sıklığı ve süresi
®
Önerilen toplam günlük NEXAVAR dozu, günde iki kez 400 mg (2 x 2 tablet) tablet şeklinde alınır.
Tedavi, hasta artık tedaviden daha fazla klinik yarar görmeyinceye veya kabul edilemez ölçüde yan etki ortaya çıkıncaya kadar sürdürülmelidir.
Uygulama şekli
NEXAVAR® oral uygulama içindir ve bir bardak su ile yutulmalıdır. Tabletler aç karına ya da düşük veya orta yağlı bir öğün ile birlikte alınabilir.
Doz titrasyonu, doz ayarlaması, özel izleme önerileri:
4.4. Özel kullanım uyarıları ve önlemleri
Deri toksisitesi durumunda önerilen doz modifikasyonları Tablo 1’de özetlenmiştir.
Tablo 1: Deri Toksisitesi Durumunda Önerilen Doz Modifikasyonları
Derece | Ortaya çıkı | NEXAVAR® doz modifikasyonu |
Derece 1 | Herhangi bir zaman | Derhal destekleyici önlemler alınır ve NEXAVAR® tedavisine devam edilir. |
Derece 2 | İlk olay | Derhal destekleyici önlemler alınır ve NEXAVAR® dozu 28 gün süre ile günde 400 mg’ a düşürülür. Doz azaltımından sonra toksisite derece 0-1’e gerilerse, 28 gün sonra sorafenib dozu artırılarak tam doza geçilir. Doz azaltımına karşın toksisite derece 0-1’e gerilemezse, en az 7 gün süreyle, toksisite derece 0-1’e dönünceye kadar NEXAVAR® tedavisine ara verilir. Aradan sonra tedaviye yeniden başlarken, NEXAVAR® 28 gün süreyle günde bir kez 400 mg’lık azaltılmış doz şeklinde uygulanır. Doz azaltımıyla toksisite derece 0-1’de tutulabilirse, 28 gün sonra NEXAVAR® dozu artırılarak tam doza geçilir. |
İkinci veya üçüncü kez ortaya çıkış | İlk ortaya çıkıştaki gibi davranılır, ancak NEXAVAR® tedavisi yeniden başlatıldıktan sonra, sürekli olarak azaltılmış günlük 400 mg dozu uygulanır. | |
Dördüncü kez | NEXAVAR® tedavisini sonlandırıp sonlandırmama kararı, klinik fayda ve hasta tercihi temellerinde alınmalıdır. | |
Derece 3 | İlk olay | Derhal destekleyici önlemler alınır ve en az 7 gün süreyle, toksisite derece 0-1’e gerileyinceye kadar NEXAVAR® tedavisine ara verilir. Aradan sonra tedaviye yeniden başlarken, NEXAVAR® 28 gün süreyle günde bir kez 400 mg’lık azaltılmış doz şeklinde uygulanır. Doz azaltımıyla toksisite derece 0-1’de tutulabilirse, 28 gün sonra NEXAVAR® dozu artırılarak tam doza geçilir. |
İkinci kez | İlk ortaya çıkıştaki gibi davranılır, ancak NEXAVAR® tedavisi yeniden başlatıldıktan sonra, sürekli olarak azaltılmış günlük 400 mg dozu uygulanır. | |
Üçüncü kez | NEXAVAR® tedavisini sonlandırıp sonlandırmama kararı, klinik fayda ve hasta tercihi temellerinde alınmalıdır. |
Özel popülasyonlara ilişkin ek bilgiler Böbrek/ karaciğer yetmezliği:
Hafif, orta dereceli ya da diyaliz gerektirmeyen şiddetli böbrek yetmezliği olan hastalarda doz ayarlaması gerekli değildir. Sorafenib diyalize girmekte olan hastalarda incelenmemiştir (bkz. Farmakokinetik özellikler - Özel popülasyonlara ilişkin ek bilgiler - Böbrek yetmezliği).
Böbrek fonksiyon bozukluğu riski olan hastalarda sıvı dengesinin ve elektrolitlerin izlenmesi önerilir.
Child-Pugh A veya B karaciğer yetmezliği olan hastalarda doz ayarlaması gerekli değildir. Sorafenib, Child-Pugh C karaciğer yetmezliği olan hastalarda incelenmemiştir (bkz. Farmakokinetik özellikler - Özel popülasyonlara ilişkin ek bilgiler - Karaciğer yetmezliği).
Pediyatrik popülasyon:
Sorafenibin çocuklardaki güvenliliği ve etkililiği belirlenmemiştir.
Geriyatrik popülasyon:
Yaşlı hastalarda (65 yaş üzeri) doz ayarlaması gerekli değildir.
Diğer:
4.3. Kontrendikasyonlar
4.4. Özel kullanım uyarıları ve önlemleri
Gebelik:
Kadınlar tedavi altında iken gebe kalmaktan kaçınmalıdır.
Çocuk doğurma potansiyeline sahip kadınlara fetus üzerindeki, ağır malformasyon (teratojenisite), büyüme geriliği ve fetal ölüm (embriyotoksisite) gibi potansiyel tehlikeler bildirilmelidir.
Sorafenib gebelik sırasında kullanılmamalıdır. Doktorlar bu ilacın kullanımını, sadece potansiyel yararları fetus üzerindeki potansiyel riskleri haklı çıkarıyorsa gündeme getirebilirler.
Multikinaz inhibisyonu için öne sürülen mekanizma ve klinik dozun oldukça altındaki miktarlara maruz kalma sonrasında hayvanlarda görülen çok sayıdaki advers etkiye dayanılarak, sorafenibin, gebe bir kadına verildiğinde fetusta zarara neden olacağı varsayılmalıdır.
Sorafenib tedavisi sırasında emzirmeye son verilmelidir.
4.6. Gebelik ve laktasyon
ve klinik öncesi güvenlilik verileri bölümleri).
Dermatolojik toksisiteler:
El-ayak deri reaksiyonu (palmar-plantar eritrodisestezi) ve döküntü sorafenibe bağlı en yaygın advers ilaç reaksiyonlarını temsil etmektedir. Döküntü ve el-ayak deri reaksiyonu sıklıkla CTC (National Cancer Institute Common Toxicity Criteria) derece 1 ve 2’dir ve genel olarak sorafenib tedavisinin ilk altı haftası içinde ortaya çıkmaktadır.
4.8. İstenmeyen etkiler
Hipertansiyon:
4.8. İstenmeyen etkiler
Hemoraji:
4.8. İstenmeyen etkiler
Varfarin:
Sorafenib tedavisinde iken varfarin alan bazı hastalarda, sık olmayan kanama olayları ya da Uluslararası Normalize Oran (INR) değerlerinde yükselmeler bildirilmiştir. Eş zamanlı olarak varfarin almakta olan hastalar, protrombin zamanında uzama, INR ve klinik kanama epizodları için düzenli şekilde izlenmelidir (bkz. İstenmeyen etkiler).
Yara iyileşmesi komplikasyonları:
Sorafenibin yara iyileşmesi üzerindeki etkisi konusunda randomize bir çalışma yürütülmemiştir. Majör cerrahi girişim geçirecek hastalarda bir önlem olarak sorafenib tedavisinin geçici olarak durdurulması önerilir. Majör cerrahi girişim sonrasında tedaviye yeniden başlama zamanı konusundaki klinik deneyim kısıtlıdır. Bu nedenle, majör cerrahi girişim sonrasında sorafenib tedavisine tekrar başlama kararı yara iyileşmesinin yeterliliğine yönelik klinik yargıya dayandırılmalıdır.
QT aralığının uzaması:
®
NEXAVAR ’ın ventriküler aritmiler açısından riskin artmasına neden olabilecek bir durum olan QT/QTc aralığını uzattığı gösterilmiştir (Bakınız Farmakolojik Özellikler). Sorafenib, konjenital uzun QT sendromu olan, yüksek kümülatif dozda antrasiklin tedavisi gören, bazı antiaritmik ilaçları veya QT uzamasına neden olan diğer tıbbi ürünleri kullanan hastalar gibi QTc uzaması olan veya gelişebilecek hastalar ile hipokalemi, hipokalsemi veya hipomagnezemi gibi elektrolit bozuklukları olan hastalarda dikkatli kullanılmalıdır. Bu hastalarda
NEXAVAR® kullanımı sırasında elektrokardiyogram ve elektrolit ölçümü (magnezyum, kalsiyum, potasyum) ile periyodik izlem göz önünde bulundurulmalıdır.
Kardiyak iskemi ve/veya enfarktüs:
Çalışma 11213’te, tedaviye bağlı kardiyak iskemi/enfarktüs olayları sorafenib grubunda (%4.9), plasebo grubuna (%0.4) kıyasla daha yüksektir. Çalışma 100554’te, tedaviye bağlı kardiyak iskemi/enfarktüs olaylarının insidansı sorafenib grubunda %2.7 iken, plasebo grubunda %1.3’tür. Stabil olmayan koroner arter hastalığı öyküsü olan ya da yakın zamanda miyokard enfarktüsü geçiren hastalar bu çalışmanın dışında bırakılmıştır. Kardiyak iskemi ve/veya enfarktüs geçiren hastalarda sorafenibe geçici olarak ya da tamamen son verilmesi gündeme getirilmelidir (bkz. İstenmeyen etkiler, Farmakodinamik özellikler -Klinik etkinlik).
Gastrointestinal perforasyon:
Gastrointestinal perforasyon sorafenib almakta olan hastalarda seyrek görülen bir advers olaydır. Hastaların %1’inden daha azında bildirilmiştir. Olguların bazılarında bu durum, görünür bir intraabdominal tümör ile ilişkilendirilememiştir. Sorafenib tedavisine son verilmelidir (bkz. İstenmeyen etkiler).
Karaciğer yetmezliği:
Child-Pugh C (şiddetli) karaciğer yetmezliği olan hastalara ilişkin veri bulunmamaktadır. Sorafenib başlıca karaciğer yoluyla elimine edildiği için şiddetli karaciğer yetmezliği olan hastalarda ilaca sistemik maruz kalma artabilir (bkz. Farmakokinetik özellikler).
İlaç etkileşimleri:
UGT1A yolu: Sorafenib, esas olarak UGT1A1 yoluyla metabolize veya elimine edilen bileşikler (örn. irinotekan) ile birlikte uygulanırken dikkatli olunması önerilmektedir (bkz. Diğer tıbbi ürünlerle etkileşimler ve diğer etkileşim şekilleri).
Dosetaksel: Dosetaksel (75 ya da 100 mg/m ) ile birlikte sorafenib (günde iki kez 200 ya da 400 mg) uygulaması (dosetaksel uygulaması sırasında sorefenibe 3 gün ara verme şeklinde), dosetaksel EAA değerinde %36-80 artış ile sonuçlanmıştır. Sorafenib dosetaksel ile birlikte uygulanırken dikkatli olunması önerilmektedir (bkz. Diğer tıbbi ürünlerle etkileşimler ve diğer etkileşim şekilleri).
Neomisin: Eş zamanlı neomisin uygulaması sorafenibin biyoyararlanımında azalmaya neden olabilir (bkz. Diğer tıbbi ürünlerle etkileşimler ve diğer etkileşim şekilleri).
4.5. Diğer tıbbi ürünlerle etkileşimler ve diğer etkileşim şekilleri
CYP3A4 indükleyicileri:
Sorafenib ve rifampisinin sürekli birlikte uygulanmaları, sorafenib EAA değerinde ortalama %37’lik bir azalma ile sonuçlanmıştır. CYP3A4 aktivitesinin diğer indükleyicileri de (örn. St. John bitkisi olarak da bilinen Hypericum perforatum, fenitoin, karbamazepin, fenobarbital ve deksametazon), sorafenib metabolizmasını artırabilir ve böylelikle sorafenib konsantrasyonlarını azaltabilirler.
CYP3A4 inhibitörleri:
Güçlü bir CYP3A4 inhibitörü olan ketokonazol, sağlıklı erkek gönüllülere 7 gün süreyle günde bir kez uygulandığında, tek doz 50 mg sorafenibin ortalama EAA değerini değiştirmemiştir. Bu nedenle sorafenib ve CYP3A4 inhibitörleri arasında klinik farmakokinetik etkileşim olasılığı pek bulunmamaktadır.
CYP2C9 substratları:
4.4. Özel kullanım uyarıları ve önlemleri
CYP izoform-selektif substratlar:
Sırasıyla, CYP3A4, CYP2D6 ve CYP2C19 substratları olan midazolam, dekstrometorfan ve omeprazol ile eş zamanlı uygulama, 4 haftalık sorafenib uygulamasından sonra bu ajanlara sistemik maruz kalma düzeylerini değiştirmemiştir. Bu durum sorafenibin, söz konusu sitokrom P450 izoenzimlerinin inhibitörü veya indükleyicisi olmadığını göstermektedir. Ayrı bir klinik çalışmada, sorafenibin paklitaksel ile eş zamanlı uygulaması, CYP2C8 tarafından oluşturulan etkin paklitaksel metaboliti 6-OH paklitaksel maruziyetinde azalma yerine artışa neden olmuştur. Bu veriler sorafenibin in vivo CYP2C8 inhibitörü olmayabileceğini ileri sürmektedir. Farklı bir klinik çalışmada sorafenibin siklofosfamid ile eşzamanlı kullanımı siklofosfamid maruziyetinde küçük bir azalma ile sonuçlanmıştır. Ancak CYP2B6 tarafından oluşturulan siklofosfamidin etkin metaboliti 4-OH siklofosfamidin sistemik maruziyetinde bir azalma meydana gelmemiştir. Bu veriler sorafenibin, CYP2B6’nın in vivo inhibitörü olmayabileceğini işaret etmektedir.
Diğer antineoplastik ajanlar ile kombinasyon:
Sorafenib klinik çalışmalarda, gemsitabin, sisplatin, oksaliplatin, paklitaksel, karboplatin, kapesitabin, doksorubisin, irinotekan, dosetaksel ve siklofosfamid gibi çeşitli diğer anti-neoplastik ajanlar ile birlikte, bu ajanların sıklıkla kullanılan dozaj rejimleriyle kullanılmıştır. Sorafenib gemsitabin, sisplatin, oksaliplatin veya siklofosfamid farmakokinetiği üzerinde klinik olarak anlamlı bir etki göstermemiştir.
Paklitaksel/Karboplatin
Paklitaksel/karboplatin uygulaması sırasında sorafenib dozunda 3 günlük bir ara verilerek, paklitaksel (225 mg/m ) ve karboplatinin (EAA=6) sorafenible (günde iki kez <400 mg) birlikte uygulanması, paklitaksel farmakokinetik değerlerinde anlamlı hiçbir etki oluşturmamıştır.
Paklitaksel (225 mg/m2, her 3 haftada bir kez) ve karboplatinin (EAA=6) sorafenible (günde iki kez 400 mg, sorafenib dozunda bir ara olmaksızın) birlikte uygulanması, sorafenib maruziyetinde %47’lik; paklitaksel maruziyetinde %29’luk ve 6-OH paklitaksel maruziyetinde ise %50’lik bir artış oluşturmuştur. Karboplatin farmakokinetik değerleri etkilenmemiştir.
Bu veriler, paklitaksel ve karboplatinin sorafenib dozunda 3 günlük bir ara dikkate alınarak sorafenible birlikte uygulanması durumunda doz ayarlamasına gerek olmadığını ortaya koymaktadır. Ara olmaksızın verilen eş zamanlı sorafenibi takiben sorafenib ve paklitaksel maruziyetlerinde gözlenen artışın klinik anlamı bilinmemektedir.
Kapesitabin
Eş zamanlı kapesitabin (750-1050 mg/m2-günde iki kez, her 21 günlük süre içinde 1-14.günlerde) ve sorafenib (günde iki kez 200 ya da 400 mg, sürekli-kesintisiz uygulama) uygulaması, sorafenib maruziyetinde anlamlı bir değişiklik oluşturmamış; ancak kapesitabin maruziyetinde %15-50’lik, 5-FU maruziyetinde ise %0-52’lik bir artışla sonuçlanmıştır. Sorafenible eşzamanlı uygulandığında kapesitabin ve 5-FU maruziyetlerinde gözlenen bu hafif ila orta düzeyli artışların klinik anlamı bilinmemektedir.
Doksorubisin/İrinotekan
Sorafenib ile eş zamanlı uygulama, doksorubisinin EAA değerinde %21’lik bir artışla sonuçlanmıştır. Aktif metaboliti SN-38’in daha sonra UGT1A1 yoluyla metabolize olduğu irinotekan ile birlikte uygulandığında, SN-38’in EAA değerinde %67-120, irinotekanın EAA değerinde ise %26-42 artış vardır. Bu bulguların klinik önemi bilinmemektedir (bkz. Özel kullanım uyarıları ve önlemleri).
Dosetaksel
Dosetaksel (21 günde bir uygulanan 75 ya da 100 mg/m ) ile sorafenibin (21 günlük siklusun 2. gününden 19. gününe kadar, günde iki kez 200 ya da 400 mg; dosetaksel uygulaması sırasında sorefenibe 3 gün ara verme şeklinde) birlikte uygulanması, dosetakselin EAA değerinde %36-80 ve dosetakselin Cmaks düzeyinde %16-32 artış ile sonuçlanmıştır. Sorafenib ile dosetakselin birlikte uygulanması sırasında dikkatli olunması önerilmektedir (bkz. Özel kullanım uyarıları ve önlemleri).
Diğer ajanlarla kombinasyon
Neomisin
4.6. Gebelik ve laktasyon
Genel tavsiye: Gebelik kategorisi D’dir.
Çocuk doğurma potansiyeli bulunan kadınlar/ Doğum kontrolü (Kontrasepsiyon):
Sorafenibin gebelik ve/veya fetüs/yenidoğan üzerinde zararlı farmakolojik etkileri bulunmaktadır. NEXAVAR® gebelik döneminde kullanılmamalıdır. Hayvanlar üzerinde yapılan araştırmalar sorafenibin teratojenik ve embriyotoksik olduğunu göstermiştir. Çocuk doğurma potansiyeli olan kadınlar tedavi süresince ve tedavinin ardından en az 2 haftaya kadar, etkili doğum kontrolü uygulamak zorundadırlar (bkz. Özel kullanım uyarıları ve önlemleri, Klinik öncesi güvenlilik verileri).
Gebelik dönemi:
Sorafenib kullanan gebe kadınlar üzerinde yürütülmüş yeterli ve iyi kontrollü çalışmalar bulunmamaktadır. Hayvanlarda yürütülen çalışmalarda malformasyonları içeren reprodüktif toksisite gösterilmiştir (bkz. Özel kullanım uyarıları ve önlemleri). Sıçanlarda sorafenib ve metabolitlerinin plasentaya geçtikleri bulunmuştur ve sorafenibin fetusta anjiyojenezi inhibe etmesi beklenmektedir.
Kadınlar tedavi altında iken gebe kalmaktan kaçınmalıdır. Çocuk doğurma potansiyeline sahip kadınlara fetüs üzerindeki ağır malformasyon (teratojenisite), büyüme geriliği ve fetal ölüm (embriyotoksisite) gibi potansiyel tehlikeler bildirilmelidir.
Sorafenib gebelik sırasında kullanılmamalıdır. Doktorlar bu ilacın kullanımını, sadece potansiyel yararları fetüs üzerindeki potansiyel riskleri haklı çıkarıyorsa gündeme getirebilirler (bkz. Özel kullanım uyarıları ve önlemleri, Klinik öncesi güvenlilik verileri).
Laktasyon dönemi:
Sorafenibin insan sütü ile atılıp atılmadığı bilinmemektedir.
Hayvanlar üzerinde yapılan çalışmalar sorafenib ve/veya metabolitlerinin süt ile
atıldığını göstermektedir.
Çoğu ilacın insanlarda süte geçmesi nedeniyle ve sorafenibin bebekler üzerindeki etkileri incelenmediğinden, kadınlar sorafenib tedavisi sırasında emzirmeyi bırakmalıdır.
Üreme yeteneği / Fertilite:
4.7. Araç ve makine kullanımı üzerindeki etkiler
NEXAVAR® ile araç ya da makine kullanma yetileri üzerindeki etkilerine yönelik çalışma yapılmamıştır. NEXAVAR®’ın araç ya da makine kullanma yetilerini etkilediğine ilişkin bir veri bulunmamaktadır.
4.8. İstenmeyen etkiler
En yaygın advers reaksiyonlar diyare, döküntü, alopesi ve el-ayak deri reaksiyonu (MedDRA’da palmar-plantar eritrodisestezi sendromuna karşılık gelir) olmuştur.
Tablo 2: Herhangi bir tedavi grubunda hastaların en az %5’inde bildirilen advers reaksiyonlar - Çalışma 11213, renal hücreli karsinoma (bkz. Çalışma 11213)
_
NEXAVAR®, n = 451 | Plasebo, n = 451 | |||||
Tüm Dereceler | Derece 3 | Derece 4 | Tüm Dereceler | Derece 3 | Derece 4 | |
Metabolizma ve beslenme hastalıkları | ||||||
Anoreksi | %9 | <%1 | %0 | %5 | <%1 | %0 |
Sinir sistemi hastalıkları | ||||||
Baş ağrısı | %6 | %0 | %0 | %3 | %0 | %0 |
Vasküler hastalıkları | ||||||
Hipertansiyon | %12 | %2 | <%1 | %1 | <%1 | %0 |
Yüz ve boyunda kızarma | %6 | %0 | %0 | %2 | %0 | %0 |
Gastrointestinal hastalıkları | ||||||
Diyare | %38 | %2 | %0 | %9 | <%1 | %0 |
Bulantı | %16 | <%1 | %0 | %12 | <%1 | %0 |
Kusma | %10 | <%1 | %0 | %6 | <%1 | %0 |
Konstipasyon | %6 | %0 | %0 | %3 | %0 | %0 |
Deri ve derialtı dokusu hastalıkları | ||||||
Döküntü | %28 | <%1 | %0 | %9 | <%1 | %0 |
Alopesi | %25 | <%1 | %0 | %3 | %0 | %0 |
El-ayak deri reaksiyonu ** | %19 | %4 | %0 | %3 | %0 | %0 |
Pruritus | %17 | <%1 | %0 | %4 | %0 | %0 |
Eritem | %15 | %0 | %0 | %4 | %0 | %0 |
Deri kuruluğu | %11 | %0 | %0 | %2 | %0 | %0 |
Deride eksfolyasyon | %7 | <%1 | %0 | %2 | %0 | %0 |
Kas-iskelet bozuklukları, ba | ||||||
Artralji | %6 | <%1 | %0 | %3 | %0 | %0 |
Ekstremite ağrısı | %6 | <%1 | %0 | %3 | %0 | %0 |
Genel bozukluklar ve uygulama bölgesine ili | ||||||
Yorgunluk | %15 | %2 | %0 | %12 | <%1 | %0 |
Asteni | %9 | <%1 | %0 | %4 | <%1 | %0 |
Tablo 3: Herhangi bir tedavi grubunda hastaların en az %5’inde bildirilen advers reaksiyonlar - Çalışma 100554, hepatoselüler karsinoma (bkz. Çalışma 100554)
__
NEXAVAR®, n = 297 | Plasebo, n = 302 | |||||
Tüm Dereceler | Derece 3 | Derece 4 | Tüm Dereceler | Derece 3 | Derece 4 | |
Metabolizma ve beslenme hastalıkları | ||||||
Anoreksi | %11 | <%1 | %0 | %3 | <%1 | %0 |
Gastrointestinal hastalıklar | ||||||
Diyare | %39 | %8 | %0 | %11 | %2 | %0 |
Bulantı | %11 | <%1 | %0 | %8 | %1 | %0 |
Abdominal ağrı | %7 | %2 | %0 | %3 | <%1 | %0 |
Kusma | %5 | %1 | %0 | %3 | <%1 | %0 |
Deri ve derialtı dokusu hastalıkları | ||||||
El-ayak deri reaksiyonu** | %18 | %7 | %0 | %2 | %0 | %0 |
Alopesi | %14 | %0 | %0 | %2 | %0 | %0 |
Döküntü | %11 | <%1 | %0 | %8 | %0 | %0 |
Pruritus | %8 | %0 | %0 | %7 | <%1 | %0 |
Deri kuruluğu | %8 | %0 | %0 | %4 | %0 | %0 |
Genel bozukluklar ve uygulama bölgesine ili | ||||||
Yorgunluk | %17 | %2 | <%1 | %13 | %3 | <%1 |
Asteni | %6 | %1 | <%1 | %2 | <%1 | %0 |
Ara | ||||||
Kilo kaybı | %9 | %2 | %0 | <%1 | %0 | %0 |
Solunum, gö | ||||||
Ses kısıklığı | %5 | %0 | %0 | <%1 | %0 | %0 |
Klinik çalışmalar sırasında meydana gelen ya da pazarlama sonrası kullanım süresince saptanan advers reaksiyonlar, aşağıda sistem-organ sınıfı (MedDRA) ve sıklık derecesine göre listelenmektedir. Sıklık dereceleri şu şekilde tanımlanmaktadır; çok yaygın (>1/10), yaygın (>1/100, <1/10), yaygın olmayan (>1/1.000, <1/100), seyrek (>1/10.000 ila <1/1.000), çok seyrek (<1/10.000), bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor).
Her sıklık grubu içinde, istenmeyen etkiler azalan şiddet derecesine göre sıralanmıştır.
Enfeksiyonlar ve enfestasyonlar
Yaygın olmayan: Folikülit, enfeksiyon
Kan ve lenf sistemi hastalıkları
Çok yaygın: Lenfopeni
Yaygın: Lökopeni, nötropeni, anemi, trombositopeni
Bağışıklık sistemi hastalıkları
Yaygın olmayan: Aşırı duyarlılık reaksiyonları (deri reaksiyonları ve ürtiker dahil) Bilinmiyor: Anjiyoödem, anafilaktik reaksiyon
Endokrin hastalıkları
Yaygın olmayan: Hipotiroidizm, hipertiroidizm
Metabolizma ve beslenme hastalıkları
Çok yaygın: Hipofosfatemi Yaygın: Anoreksi
Yaygın olmayan: Hiponatremi, dehidrasyon
Psikiyatrik hastalıklar
Yaygın: Depresyon
Sinir sistemi hastalıkları
Yaygın: Periferik duysal nöropati
Yaygın olmayan: Geri dönüşümlü posterior lökoensefalopati*
Kulak ve içkulak hastalıkları
Yaygın: Tinnitus
Kardiyak hastalıklar
Yaygın: Konjestif kalp yetmezliği*, miyokard iskemisi ve/veya enfarktüsü* Seyrek: QT uzaması
Vasküler hastalıklar
Çok yaygın: Hemoraji (gastrointestinal,* solunum yolu* ve serebral hemoraji*
dahil), hipertansiyon
Yaygın olmayan: Hipertansif kriz*
Solunum, göğüs bozuklukları ve mediyastinal hastalıklar
Yaygın: Ses kısıklığı
Yaygın olmayan: Rinore, interstisyel akciğer hastalığı benzeri olaylar (pnömonit, radyasyon pnömoniti, akut solunum güçlüğü, interstisyel pnömoni, pulmonit ve akciğer inflamasyonu bildirimleri de dahil olmak üzere).
Gastrointestinal hastalıklar
Çok yaygın: Diyare, bulantı, kusma
Yaygın: Konstipasyon, stomatit (ağız kuruluğu ve glossodini dahil), dispepsi, disfaji
Yaygın olmayan: Gastroözofajiyal reflü hastalığı, pankreatit, gastrit, gastrointestinal perforasyonlar*
Hepatobiliyer hastalıklar
Yaygın olmayan: Bilirübin artışı ve sarılık, kolesistit, kolanjit Bilinmiyor: İlaç nedenli hepatit*
Deri ve derialtı dokusu hastalıkları
Çok yaygın: Döküntü, alopesi, el-ayak deri reaksiyonu**, pruritus, eritem Yaygın: Deri kuruluğu, eksfolyatif dermatit, akne, deri deskuamasyonu Yaygın olmayan: Egzama, eritema multiforme, deride keratoakantomalar/ skuamöz hücreli kanser
Bilinmiyor: Radyasyon dermatiti, Stevens-Johnson sendromu
Kas-iskelet bozuklukları, bağ dokusu ve kemik hastalıkları
Yaygın: Artralji, miyalji
Renal ve genitoüriner bozukluklar
Yaygın: Böbrek yetmezliği
Üreme sistemi ve meme hastalıkları
Yaygın: Erektil disfonksiyon Yaygın olmayan: Jinekomasti
Genel bozukluklar ve uygulama yerine özgü tablolar
Çok yaygın: Yorgunluk, ağrı (ağız, abdominal, kemik, tümör ağrısı ve baş ağrısı dahil)
Yaygın: Asteni, ateş, grip benzeri hastalık
Araştırmalar
Çok yaygın: Amilaz artışı, lipaz artışı Yaygın: Kilo kaybı, transaminazlarda geçici artış
Yaygın olmayan: Kan alkalen fosfatazında geçici artış, anormal INR (Uluslararası Normalizasyon Oranı), protrombin düzeyinde anormallik
* Bu advers reaksiyonlar hayatı tehdit edici ya da ölümcül sonuç verebilir. Bu gibi olaylar ya seyrek ya da seyrekten daha az sıklıktadır. **MedDRA’da palmar plantar eritrodisestezi.
Bazı advers ilaç reaksiyonları ile ilgili ilave bilgi
Konjestif Kalp Yetmezliği - Yürütülen klinik çalışmalarda, konjestif kalp yetmezliği sorafenib tedavisi gören hastaların (n=2.276) %1.9’unda advers olay olarak bildirilmiştir. Çalışma 11213’te (RCC) konjestif kalp yetmezliğiyle uyumlu advers olaylar NEXAVAR® tedavisi gören hastaların %1.7’sinde ve plasebo grubunun %0.7’sinde bildirilmiştir. Çalışma 100554’de (HCC), NEXAVAR® tedavisi görenlerin %0.99’unda, plasebo kullananların ise %1.1’inde bu olaylar bildirilmiştir.
İleri Küçük Hücre Dışı Akciğer Kanseri (KHDAK) olan hastaların ilk basamak tedavisi olarak sorafenibin ikili platinyum-bazlı kemoterapilerle (karboplatin/paklitaksel ve ayrı ayrı gemsitabin/sisplatin) kombine kullanımına karşılık sadece ilgili ikili platinyum-bazlı kemoterapinin güvenlilik ve etkililiğinin karşılaştırıldığı iki randomize plasebo kontrollü çalışma, genel sağkalımın iyileşmesi olan birincil sonlanım noktasına ulaşamamıştır. Güvenlilik olayları genellikle daha önce bildirilen olaylarla uyumludur. Ancak, iki çalışmada da tek başına ikili platinyum-bazlı kemoterapi (karboplatin/paklitaksel ve ayrı ayrı gemsitabin/sisplatin) tedavisiyle karşılaştırıldığında, sorafenib ve ikili platinyum-bazlı kemoterapi tedavisi uygulanan skuamöz hücreli akciğer kanseri olan bir hasta alt grubunda daha yüksek mortalite izlenmiştir (HR 1.22, %95 GA 0.821.80). Bu bulgulara ilişkin kesin bir gerekçe tanımlanmamıştır.
Güvenlilik aynı zamanda bir faz II çalışma havuzunda da değerlendirilmiştir. Bu toplu veriler, NEXAVAR® ile tedavi edilen 638 hastaya aittir ve bunların arasında 202 hastada renal hücreli karsinoma, 137 hastada hepatoselüler karsinoma ve 299 hastada başka kanser türleri bulunmaktadır. Bu toplu veriler içinde NEXAVAR® tedavisindeki hastalarda en sık bildirilen ilaca bağlı advers olaylar; döküntü (%38), diyare (%37), el-ayak deri reaksiyonu (%35) ve yorgunluk (%33) olmuştur. NEXAVAR® tedavisindeki hastalarda CTC (v 2.0) Derece 3 ve 4 ilaca bağlı advers olaylar, sırasıyla %37 ve %3’dür.
RCC hastalarında laboratuar test anormallikleri (Çalışma 11213): Lipaz ve amilaz düzeylerinde artış sık olarak bildirilmiştir. Çalışma 11213’te, CTCAE derece 3 ya da 4 lipaz artışları, sorafenib grubundaki hastaların %12’sinde bildirilirken, plasebo grubundaki hastaların %7’sinde bildirilmiştir. CTCAE derece 3 ya da 4 amilaz artışları, sorafenib grubundaki hastaların %1’inde bildirilirken, plasebo grubundaki hastaların %3’ünde bildirilmiştir. Çalışma 1’de klinik pankreatit, sorafenib tedavisindeki 451 hastanın 2’sinde (CTCAE derece 4) ve plasebo grubundaki 451 hastanın 1’inde (CTCAE derece 2) bildirilmiştir.
Hipofosfatemi sık rastlanan laboratuar anomalisidir, sorafenib tedavisindeki hastaların %45’inde, plasebo hastalarının %11’inde gözlenmiştir. CTCAE derece 3 hipofosfatemi (1-2 mg/dl), sorafenib ile tedavi edilen hastaların %13’ünde ve plasebo grubundaki hastaların %3’ünde ortaya çıkmıştır. CTCAE derece 4 hipofosfatemi (< 1 mg/dl) olgusu sorafenib ve plasebo kollarında bildirilmemiştir. Sorafenib ile ilişkili hipofosfateminin etiyolojisi bilinmemektedir.
CTCAE derece 3 ya da 4 olaylar, lenfopeni için sorafenib tedavisindeki hastaların %13’ünde ve plasebo hastalarının %7’sinde, nötropeni için sorafenib tedavisindeki hastaların %5 ve plasebo hastalarının %2’sinde, anemi için sorafenib tedavisindeki hastaların %2 ve plasebo hastalarının %4’ünde, ve trombositopeni için sorafenib tedavisindeki hastaların %1 ve plasebo hastalarının %0’ında bildirilmiştir.
HCC hastalarında laboratuar anormallikleri (Çalışma 100554): Lipaz artışı NEXAVAR® ile tedavi edilen hastaların %40’ında gözlenirken, plasebo grubundaki hastaların %37’sinde gözlenmiştir. CTCAE derece 3 ya da 4 lipaz artışları, her iki grupta da hastaların %9’unda ortaya çıkmıştır. Amilaz artışları, NEXAVAR® ile tedavi edilen hastaların %34’ünde gözlenirken, plasebo grubundaki hastaların %29’unda gözlenmiştir. CTCAE derece 3 ya da 4 amilaz artışları, her iki grupta da hastaların %2’sinde bildirilmiştir. Lipaz ve amilaz artışlarının çoğu geçicidir ve olguların büyük çoğunluğunda NEXAVAR® tedavisine ara verilmemiştir. Klinik pankreatit NEXAVAR® tedavisindeki 297 hastanın 1’inde bildirilmiştir (CTCAE derece 2).
Hipofosfatemi sık rastlanan bir laboratuar bulgusudur, NEXAVAR® ile tedavi edilen hastaların %35’inde, plasebo hastalarının %11’inde gözlenmiştir. CTCAE derece 3 hipofosfatemi (1-2 mg/dL), NEXAVAR® ile tedavi edilen hastaların %11’inde ve plasebo grubundaki hastaların %2’sinde ortaya çıkmıştır; plasebo grubunda bildirilen 1 CTCAE derece 4 hipofosfatemi (<1 mg/dL) olgusu bulunmaktadır. NEXAVAR® ile ilişkili hipofosfateminin etiyolojisi bilinmemektedir.
Karaciğer fonksiyon testlerindeki artışlar, çalışmanın 2 kolu arasında karşılaştırılabilir niteliktedir. AST artışları NEXAVAR® ile tedavi edilen hastaların %94’ü ve plasebo hastalarının %91’inde gözlenmiştir. CTCAE derece 3 ya da 4 AST artışları, NEXAVAR® ile tedavi edilen hastaların %16’sında ve plasebo grubundaki hastaların %17’sinde bildirilmiştir. ALT artışları NEXAVAR® ile tedavi edilen hastaların %69’unda ve plasebo hastalarının %68’inde gözlenmiştir. CTCAE derece 3 ya da 4 ALT artışları, NEXAVAR® ile tedavi edilen hastaların %3’ünde ve plasebo tedavisindeki hastaların %8’inde bildirilmiştir. Bilirübin artışları, NEXAVAR® ile tedavi edilen hastaların %47’sinde ve plasebo hastalarının %45’inde gözlenmiştir. CTCAE derece 3 ya da 4 bilirübin artışları NEXAVAR® ile tedavi edilen hastaların %10’unda ve plasebo tedavisindeki hastaların %11’inde bildirilmiştir. Hipoalbüminemi, NEXAVAR® ile tedavi edilen hastaların %59’unda ve plasebo hastalarının %47’sinde gözlenmiştir; her iki grupta da CTCAE derece 3 ya da 4 hipoalbüminemi gözlenmemiştir.
Alkalen fosfataz artışları, NEXAVAR® ile tedavi edilen hastaların %82.2’sinde ve plasebo hastalarının %82.5’inde gözlenmiştir. CTCAE derece 3 alkalen fosfataz artışları, NEXAVAR® ile tedavi edilen hastaların %6.2’sinde ve plasebo tedavisindeki hastaların %8.2’sinde bildirilmiştir; her iki grupta da hiç CTCAE derece 4 alkalen fosfataz artışı gözlenmemiştir.
INR artışları, NEXAVAR® ile tedavi edilen hastaların %42’sinde ve plasebo hastalarının %34’ünde gözlenmiştir. CTCAE derece 3 INR artışları NEXAVAR® ile tedavi edilen hastaların %4’ünde ve plasebo hastalarının %2’sinde bildirilmiştir; her iki grupta da CTCAE derece 4 INR yükselmesi bulunmamaktadır.
Lenfopeni, NEXAVAR® ile tedavi edilen hastaların %47’sinde ve plasebo hastalarının %42’sinde gözlenmiştir. CTCAE Derece 3 ya da 4 lenfopeni, her bir gruptaki hastaların %6’sında bildirilmiştir. Nötropeni NEXAVAR® ile tedavi edilen hastaların %11’inde ve plasebo hastalarının %14’ünde gözlenmiştir. CTCAE derece 3 ya da 4 nötropeni, her bir gruptaki hastaların %1’inde bildirilmiştir.
Anemi, NEXAVAR® ile tedavi edilen hastaların %59’unda ve plasebo hastalarının %64’ünde gözlenmiştir. CTCAE Derece 3 ya da 4 anemi, her bir gruptaki hastaların %3’ünde bildirilmiştir.
4.9. Doz aşımı ve tedavisi
NEXAVAR® doz aşımı için spesifik bir tedavi bulunmamaktadır.
Klinik olarak incelenen en yüksek sorafenib dozu günde iki kez 800 mg’dır. Bu dozda gözlenen advers reaksiyonlar başlıca diyare ve dermatolojik olaylar olmuştur.
®
Bir doz aşımı kuşkusu durumunda, NEXAVAR® uygulaması durdurulmalı ve destekleyici tedavi başlatılmalıdır.
5. FARMAKOLOJİK ÖZELLİKLER
5.1. Farmakodinamik özellikler
Farmakoterapötik grup: Protein kinaz inhibitörü ATC kodu: L01XE05
Etki mekanizması
Sorafenib in vitro tümör hücresi proliferasyonunu azaltan bir multikinaz inhibitörüdür.
Sorafenibin çok sayıda hücre içi (c-CRAF, BRAF ve mutant BRAF) ve hücre yüzeyi kinazlarını (KIT, FLT-3, RET, VEGFR-1, VEGFR-2, VEGFR-3, ve PDGFR-B) inhibe ettiği gösterilmiştir. Bu kinazlardan bazılarının tümör hücresi sinyal iletimi, anjiyogenez ve apoptozda rol aldığı düşünülmektedir. Sorafenib immünokompromize farelerde, insan hepatoselüler karsinoma ve renal hücreli karsinomada ve çeşitli başka insan tümör ksenogreftlerinde tümör büyümesini inhibe etmiştir. İnsan hepatoselüler ve renal hücreli karsinoma modellerinde, tümör anjiyogenezinde azalma ve tümör apopitozunda artışlar görülmüştür. Ayrıca, insan hepatoselüler karsinoma modelinde tümör hücresi sinyal iletiminde azalma görülmüştür.
Klinik etkililik
Hepatoselüler karsinoma
Çalışma 100554, hepatoselüler karsinomalı 602 hasta üzerinde yürütülen, uluslararası, çok-merkezli, randomize, çift-kör, plasebo-kontrollü bir Faz III araştırmadır. Bu çalışmanın birincil sonlanım noktası genel sağkalım (OS), ikincil
sonlanım noktası ise progresyona kadar geçen süre (TTP)’dir.
®
NEXAVAR ve plasebo grupları arasında demografik özellikler ve başlangıç dönemi hastalık özellikleri, aşağıdaki parametreler yönüyle benzerdir; yaş, cinsiyet, ırk, performans statüsü, etiyoloji (hepatit B, hepatit C ve alkolik karaciğer hastalığı dahil), TNM evresi (evre I: <%1 vs <%1; evre II: %10.4 vs %8.3; evre III: %37.8 vs %43.6; evre IV: %50.8 vs %46.9), makroskopik vasküler invazyon ve ekstrahepatik tümör yayılımı olmaması (%30.1 vs %30.0), ve BCLC evresi (evre B: %18.1 vs %16.8; evre C: %81.6 vs %83.2; evre D: <%1 vs %0). Karaciğer fonksiyonu Child-Pugh statüsü, NEXAVAR® ve plasebo grupları arasında benzerdir (A: %95 vs %98; B: %5 vs %2). Çalışmada Child-Pugh C karaciğer disfonksiyonu olan sadece bir hasta tedavi edilmiştir. Önceden uygulanan tedaviler, cerrahi rezeksiyon prosedürleri (%19.1 vs %20.5), lokorejyonal tedaviler (radyofrekans ablasyonu, perkütan etanol enjeksiyonu ve
transarteryel kemoembolizasyon; %38.8 vs %40.6), radyoterapi (%4.3 vs %5.0) ve sistemik tedavidir (%3.0 vs %5.0).
Çalışma, genel sağkalım üzerine yapılması planlanmış olan bir ara analiz değerlendirmesi sonucunda, daha önceden belirlenmiş olan etkililik eşik değeri aşıldığı için durdurulmuştur. Bu OS analizinde NEXAVAR® için plaseboya karşı, genel sağkalıma yönelik istatistiksel olarak anlamlı bir avantajın bulunduğu gösterilmiştir (Tehlike oranı (TO): 0.69, p= 0.00058, bkz. Tablo 4 ve Şekil 1). Bu avantaj, analiz edilen alt-grupların neredeyse tamamında tutarlılık göstermiştir. Önceden belirlenmiş katmanlaştırma (stratifikasyon) faktörleri için (ECOG statüsü, makroskopik vasküler invazyon ve/veya ekstrahepatik tümör yayılımının varlığı ya da yokluğu ve bölge) tehlike oranı değerleri, plaseboya karşı NEXAVAR®’ın lehinedir. Progresyona kadar geçen süre (TTP, bağımsız radyolojik inceleme ile) NEXAVAR® kolunda anlamlı olarak daha uzundur (TO: 0.58, p= 0.000007, bkz. Tablo 4).
Tablo 4: Çalı
ş
ma 100554: Hepatoselüler Karsinomada Etkinlik Sonuçları
Etkinlik Parametresi | NEXAVAR® | Plasebo | P değeri | TO |
(n=299) | (n=303) | (%95 GA) | ||
Genel Sağkalım [ortanca, hafta (%95 GA)] | 46.3 (40.9, 57.9) | 34.4 (29.4, 39.4) | 0.00058* | 0.69 (0.55, 0.87) |
Progresyona kadar geçen süre [ortanca, hafta (%95 GA)]1 | 24.0 (18.0, 30.0) | 12.3 (11.7, 17.1) | 0.000007 | 0.58 (0.45, 0.74) |
GA=Güven aralığı, TO=Tehlike oranı (plaseboya karşı NEXAVAR®)
* p değeri önceden belirlenmiş O’Brien Fleming durdurma sınır değeri olan 0.0077’nin altında olduğu için istatistiksel olarak anlamlı.
Ş
ekil 1: : Çalı
ş
ma 100554: Genel sa
ğ
kalım (OS) için Kaplan-Meier E
ğ
risi (ITT hasta grubu)
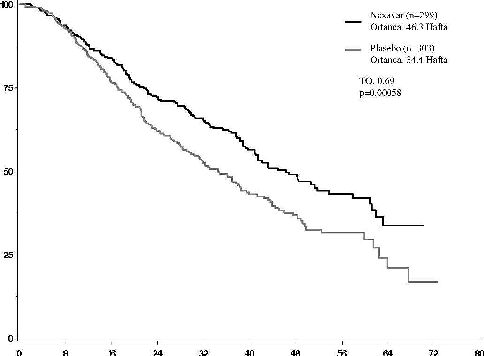
Randomizasyon sonrası haftalar
Riskli hastalar
Sorafenib 274 241 205 161 108 67 38 12 0 0
Plasebo 276 224 179 126 78 47 25 7 2 0
Renal hücreli karsinoma
NEXAVAR®’ın ilerlemiş renal hücreli karsinoma (RCC) tedavisindeki güvenlilik ve etkililik
2 randomize kontrollü klinik çalışmada incelenmiştir:
Çalışma 1 (Çalışma 11213), 903 hastanın alındığı Faz III, uluslararası, çok-merkezli, randomize, çift-kör, plasebo-kontrollü bir çalışmadır. Birincil sonlanım noktaları genel sağkalım ve progresyonsuz sağkalımdan (PFS) oluşmaktadır. Tümör yanıt oranı ikincil sonlanım noktasıdır.
Hastalar günde iki kez 400 mg NEXAVAR® (n = 451) ya da plaseboya (n = 452) randomize edilmiştir. Başlangıç dönemi demografik özellikleri ve hasta özellikleri her iki tedavi grubu arasında iyi bir dengelenme göstermiştir. Hastaların yaklaşık yarısının ECOG performans statüsü 0’dır ve hastaların yarısı düşük MSKCC (Memorial Sloan Kettering Kanser Merkezi) prognostik grubu içindedir.
220 ölüm olayına dayalı planlanmış bir sağkalım ara analizinde, sorafenib alan hastalar için plaseboya kıyasla genel sağkalımda %39 düzeyinde bir iyileşmenin bulunduğu görülmüştür. Hesaplanan tehlike oranı (sorafenib ile plaseboya kıyasla ölüm riski) 0.72’dir (%95 GA, 0.55-0.95; p=0.018. Ara analizdeki istatistiksel anlamlılık eşiği p<0.0005 değeridir).
PFS analizi, günde iki kez 400 mg NEXAVAR® (n = 384) ya da plaseboya (n = 385) randomize edilen 769 hastayı kapsamaktadır. PFS, körlemeli bağımsız radyolojik inceleme yoluyla, RECIST kriterleri kullanılarak değerlendirilmiştir. Ortanca PFS, sorafenibe randomize edilen hastalarda (167 gün) plasebo hastalarındakinin (84 gün) iki katıdır (TO= 0.44; %95 GA: 0.35-0.55; p<0.000001).
PFS üzerindeki etki aynı zamanda değişik hasta alt-gruplarında da araştırılmıştır. Alt gruplar şu özelliklere göre oluşturulmuştur; yaşın 65’in altında ya da üzerinde oluşu, ECOG PS 0 ya da 1, MSKCC prognostik risk kategorisi 1, önceki tedavinin progresif metastatik hastalık için mi, yoksa hastalığın daha erken döneminde mi uygulandığı ve tanıdan itibaren geçen sürenin 1.5 yıldan daha kısa veya daha uzun olması. Sorafenibin PFS üzerindeki etkisi, bu alt-gruplar arasında sürekli ve tutarlıdır. Yine alt-gruplarda yer alan, önceden IL-2 ya da interferon tedavisi görmemiş olan hastalarda (n=137; sorafenib alan 65 hasta ve 72 plasebo hastası), ortanca PFS sorafenib ile 172 gün iken, plasebo ile 85 gün olmuştur.
En iyi genel tümör yanıtı, araştırıcının radyolojik incelemesiyle, RECIST kriterleri doğrultusunda belirlenmiştir. Sorafenib grubunda 1 hastada (%0.2) tam yanıt, 43 hastada (%9.5) parsiyel yanıt ve 333 hastada (%73.8) stabil hastalık bulunmaktadır. Plasebo grubunda 0 hastada (%0) tam yanıt, 8 hastada (%1.8) parsiyel yanıt ve 239 hastada (%52.9) stabil hastalık vardır.
Sorafenib ile plaseboya kıyasla, böbrek kanserine özgü semptomlarda (FKSI-10) ya da sağlığa ilişkin yaşam kalitesinde genel bir kötüleşme izlenmemiştir. Tedavinin 18 ve 24. haftalarında, sorafenib alan daha fazla sayıda hastada total FKSI-10 skoru (sırasıyla %55 ve %44) ve fiziksel iyilik hali (FACT-G PWB) skorunda (sırasıyla %57 ve %47) iyileşme bildirilmiştir; plasebo grubunda ise bu oranlar, sırasıyla FKSI-10, %33 ve %21 ve FACT-G PWB %37 ve %21 olmuştur.
Çalışma 2, RCC dahil metastatik maligniteleri olan hastalarda randomize bir faz II tedavi sonlandırma araştırmasıdır. Çalışmanın birincil sonlanım noktası, randomize edilen hastalar (n= 65) arasında 24. haftada progresyonsuz sağkalım oranıdır. Progresyonsuz sağkalım, sorafenib grubunda (163 gün) plasebo grubunda olduğundan (41 gün) anlamlı olarak daha uzundur (p=0.0001, TO=0.29). Progresyon göstermeyenlerin oranı sorafenibe randomize edilen hastalar arasında (%50) plasebo hastalarındakinden (%18) anlamlı olarak daha yüksektir (p=0.0077). QT aralığının uzaması
5.2. Farmakokinetik özellikler
Genel özellikler
Sorafenib tablet uygulamasından sonra, oral çözeltiye kıyasla ortalama bağıl biyoyararlanım %38-49’dur. Sorafenibin eliminasyon yarı-ömrü 25-48 saat civarındadır. Yedi gün süreyle çoklu sorafenib dozlarının uygulanması, tek doz uygulamaya kıyasla 2.5 ile 7 katlık bir birikim ile sonuçlanır. Kararlı durum
plazma sorafenib konsantrasyonları 7 gün içinde elde edilir, ve ortalama konsantrasyonların tepe-vadi oranı 2’den düşüktür.
Emilim:
Oral uygulamayı izleyerek, sorafenib doruk plazma düzeylerine yaklaşık 3 saatte ulaşır. Orta dereceli yağ içeren bir yemek ile verildiğinde, biyoyararlanımı aç karına olduğu gibidir. Yağdan zengin bir yemek ile verildiğinde, sorafenib biyoyararlanımı aç karına uygulamaya kıyasla %29 azalmaktadır. Günde iki kez uygulanan 400 mg’ın ötesindeki dozlarda, ortalama Cmaks ve EAA değerleri, dozla orantılı düzeylerden daha düşük bir artış gösterir.
Dağılım:
Sorafenibin insan plazma proteinlerine in vitro bağlanması %99.5 düzeyindedir. Biyotransformasyon:
Sorafenib esas olarak karaciğerde metabolize olur, CYP3A4’ün aracılık ettiği oksidatif metabolizmaya girerken, aynı zamanda UGT1A9 aracılığıyla glukuronidasyona da uğrar. Sorafenib konjugatları gastrointestinal kanalda bakteriyel glukuronidaz aktivitesi tarafından parçalanabilirler, bu da konjuge olmayan ilacın tekrar emilimini sağlar. Eş zamanlı neomisin uygulaması bu süreci engelleyerek sorafenibin ortalama biyoyararlanımını %54 oranında düşürür.
Sorafenib kararlı durumda, plazmada dolaşan analitlerin yaklaşık %70-85’ini oluşturur. Sorafenibin sekiz metaboliti tanımlanmıştır, bunlardan beşi plazmada saptanmıştır. Sorafenibin plazmada dolaşan esas metaboliti, piridin N-oksit, sorafenibe benzer bir in vitro potens gösterir. Bu metabolit kararlı durumda dolaşımdaki analitlerin yaklaşık %9-16’sını oluşturur.
Eliminasyon:
Çözelti şeklinde bir sorafenib formülasyonu oral yoldan 100 mg dozda uygulandıktan sonra dozun %96’sı 14 gün içinde atılmıştır, bu miktarın %77’si feçes ile, %19’u idrarda glukuronize metabolitler şeklinde atılmıştır. Dozun %51’ini oluşturan değişmemiş haldeki sorafenib, feçeste bulunmakta, ama idrarda bulunmamaktadır.
Doğrusallık/Doğrusal olmayan durum: Sorafenib doğrusal farmakokinetik özellik gösterir.
Enzim inhibisyonu çalışmaları:
İnsan karaciğer mikrozomlarında yapılan çalışmalarda, sorafenibin CYP2C19, CYP2D6 ve CYP3A4’ün yarışmacı bir inhibitörü olduğu ortaya konulmuştur. Sırasıyla sitokrom CYP3A4, CYP2D6 ve CYP2C19 substratları olan midazolam, dekstrometorfan ve omeprazolün 4 haftalık sorafenib uygulamasını takiben eş zamanlı uygulaması bu ajanların maruziyetini değiştirmemiştir. Bu bulgu sorafenibin söz konusu sitokrom P450 izoenzimleri açısından bir inhibitor ya da indükleyici olmadığını göstermektedir
İn vitro veriler sorafenibin UGT1A1 ve UGT1A9 yolları aracılığıyla oluşan glukuronidasyonu inhibe ettiğini göstermektedir. Etkin metaboliti SN-38’in UGT1A1 yoluyla metabolize olduğu irinotekan ile sorafenibin eşzamanlı klinik uygulaması SN-38 EAA’sında %67-%120’lik bir artışa neden olmuştur. Sorafenib ile birlikte uygulandıklarında, UGT1A1 ve UGT1A9 substratlarına sistemik maruz kalma düzeyleri artabilir.
Sorafenib CYP2B6 ve CYP2C8’i, sırasıyla 6 ve 1-2 ||M düzeyindeki Ki değerleriyle in vitro inhibe eder. Sorafenibin paklitaksel ile eşzamanlı klinik uygulaması, paklitakselin CYP2C8 tarafından oluşturulan etkin metaboliti olan 6-OH maruziyetinde düşüş yerine artışa neden olmuştur. Bu veriler sorafenibin in vivo CYP2C8 inhibitörü olmayabileceğini ortaya koymaktadır. Sorafenibin siklofosfamid ile eşzamanlı kullanımı siklofosfamid maruziyetinde küçük bir azalma ile sonuçlanmıştır. Ancak CYP2B6 tarafından oluşturulan siklofosfamidin etkin metaboliti 4-OH siklofosfamidin sistemik maruziyetinde bir azalma meydana gelmemiştir. Bu veriler, sorafenibin CYP2B6’nın in vivo inhibitörü olmayabileceğini işaret etmektedir.
İnsan karaciğer mikrozomlarıyla yapılan çalışmalarda sorafenibin, 7-8 | M düzeyinde bir Ki değeriyle, CYP2C9’un yarışmacı bir inhibitörü olduğu gösterilmiştir. Sorafenibin bir CYP2C9 substratı üzerindeki olası etkisi, varfarin ile kombinasyon şeklinde sorafenib ya da plasebo almakta olan hastalarda değerlendirilmiştir. PT-INR değerlerinde başlangıç dönemine göre ortalama değişimler, sorafenib hastalarında plasebo hastalarına kıyasla daha yüksek değildir. Bu durum sorafenibin, CYP2C9’un bir in vivo inhibitörü olmayabileceğini düşündürmektedir.
CYP3A4 inhibitörlerinin etkisi:
Güçlü bir CYP3A4 inhibitörü olan ketokonazol (400 mg), sağlıklı erkek gönüllülere 7 gün süreyle günde bir kez uygulandığında, tek doz 50 mg sorafenibin ortalama EAA değerini değiştirmemiştir. Dolayısıyla, sorafenibin CYP3A4 inhibitörleriyle klinik farmakokinetik etkileşimleri olası değildir
CYP enzim indükleyicilerinin etkisi:
Kültüre insan hepatositleri üzerine sorafenib uygulandıktan sonra, CYP1A2 ve CYP3A4 aktiviteleri değişmemiştir; bu durum sorafenibin CYP1A2 ve CYP3A4 indükleyicisi olma olasılığının uzak olduğuna işaret etmektedir. Sorafenib ile rifampisinin sürekli eşzamanlı uygulaması sorafenib EAA’sında ortalama %37’lik bir azalmayla sonuçlanmıştır. CYP3A4 aktivitesine ilişkin diğer indükleyiciler (ör, sarı kantaron (St.John’s wort) olarak da bilinen Hypericum perfornatum, fenitoin, karbamazepin, fenobarbital ve deksametazon) de sorafenibin metabolizmasını artırabilir, dolayısıyla sorafenib konsantrasyonlarını azaltabilir.
Diğer anti-neoplastik ajanlarla kombinasyon:
Klinik çalışmalarda sorafenib, gembsitabin, sisplatin, oksaliplatin, paklitaksel, karboplatin, kapesitabin, doksorubisin, dosetaksel, irinotekan ve siklofosfamid de dahil olmak üzere, yaygın kullanılan doz rejimleri doğrultusunda diğer antineoplastik ajanlarla birlikte uygulanmıştır. Sorafenibin gemsitabin, sisplatin, oksaliplatin ya da siklofosfamid farmakokinetikleri üzerinde klinik olarak anlamlı hiçbir etkisi mevcut değildir.
Paklitaksel/Karboplatin:
Paklitaksel/karboplatin uygulaması sırasında sorafenib dozunda 3 günlük bir ara verilerek, paklitaksel (225 mg/m2) ve karboplatinin (EAA=6) sorafenible (günde iki kez <400 mg) birlikte uygulanması paklitaksel farmakokinetik değerlerinde anlamlı hiçbir etki oluşturmamıştır.
Paklitaksel (225 mg/m2, her 3 haftada bir kez) ve karboplatinin (EAA=6) sorafenible (günde iki kez 400 mg, sorafenib dozunda bir ara olmaksızın) birlikte uygulanması, sorafenib maruziyetinde % 47’lik; paklitaksel maruziyetinde %29’luk ve 6-OH paklitaksel maruziyetinde ise %50’lik bir artış oluşturmuştur. Karboplatin farmakokinetik değerleri etkilenmemiştir.
Bu veriler, paklitaksel ve karboplatinin sorafenib dozunda 3 günlük bir ara dikkate alınarak sorafenible birlikte uygulanması durumunda doz ayarlamasına gerek olmadığını ortaya koymaktadır. Ara olmaksızın verilen eşzamanlı sorafenibi takiben sorafenib ve paklitaksel maruziyetlerinde gözlenen artışın klinik anlamı bilinmemektedir.
Kapesitabin:
Eşzamanlı kapesitabin (750-1050 mg/m2-günde iki kez, her 21 günlük süre içinde 1-14. günlerde) ve sorafenib (günde iki kez 200 ya da 400 mg, sürekli, kesintisiz uygulama) uygulaması sorafenib maruziyetinde anlamlı bir değişiklik oluşturmamış ancak kapesitabin maruziyetinde %15-50’lik, 5-FU maruziyetinde ise %0-52’lik bir artışla sonuçlanmıştır. Sorafenible eşzamanlı uygulandığında kapesitabin ve 5-FU maruziyetlerinde gözlenen bu hafif ila orta düzeyli artışların klinik anlamı bilinmemektedir.
Doksorubisin/İrinotekan:
Sorafenib ile eşzamanlı tedavi sonucu doksorubisin EAA’sında %21’lik bir artış izlenmiştir. Etkin metaboliti SN-38’in UGT1A1 yoluyla ile metabolize olduğu irinotekanla birlikte uygulandığında ise SN-38 EAA’sında %67-120’lik, irinotekan EAA’sında %26-42’lik bir artış söz konusudur. Bu bulguların klinik anlamı bilinmemektedir (Kullanıma yönelik Özel Uyarılar ve Önlemler başlığına bakınız).
Dosetaksel:
Dosetaksel (her 21 günde bir kez uygulanan 75 ya da 100 mg/m2 ) ile birlikte 3 günlük bir doz arası ile uygulanan sorafenib (21 günlük siklusun 2. gününden 19. gününe kadar, günde iki kez 200 ya da 400 mg, dosetaksel EAA’sında %36-80’lik, Cmaks değerinde ise %16-32’lik bir artışa neden olmuştur. Sorafenibin dosetaksel ile birlikte uygulandığı durumda dikkatli olunması önerilmektedir (Kullanıma Yönelik Özel Uyarılar ve Önlemler başlığına bakınız).
Diğer ajanlarla kombinasyon:
Neomisin
Gİ florayı eradike etmek üzere kullanılan sistemik olmayan bir antimikrobiyal ajan olan neomisinin eşzamanlı uygulaması, sorafenibin enterohepatik geri dönüşümünü engellemekte (yukarıdaki kısma bakınız), sorafenib maruziyetinin azalmasına neden olmaktadır. 5 günlük bir neomisin rejiminin uygulandığı sağlıklı gönüllülerde sorafenibin ortalama biyoyararlanımı %54 oranında azalmıştır. Bu bulguların klinik anlamı henüz bilinmemektedir. Diğer antibiyotiklerin etkileri
araştırılmamıştır ancak glukuronidaz aktivitesini azaltma yetkinliklerine göre değişkenlik göstermesi olasıdır.
Hastalardaki karakteristik özellikler
Irk:
Beyaz ırk ile Asya ırkına mensup gönüllülerde farmakokinetik özellikler bakımından klinik olarak anlamlı farklılıklar bulunmamaktadır.
Cinsiyet:
Demografik verilerin analizleri, cinsiyete göre doz ayarlaması yapılmasının gerekli olmadığını göstermektedir.
Karaciğer yetmezliği:
Sorafenib esas olarak karaciğer tarafından elimine edilmektedir. Hafif (Child-Pugh A) ya da orta derecede (Child-Pugh B) karaciğer yetmezliği olan hepatoselüler karsinomu hastalarda sistemik maruz kalma düzeyleri, karaciğer bozukluğu olmayan hastalarda gözlenen aralık içindedir. Child-Pugh A ve Child-Pugh B olup hepatoselüler karsinomu olmayan hastalarda sorafenibin farmakokinetiği, sağlıklı gönüllülerin farmakokinetiği ile benzerdir. Sorafenib farmakokinetiği şiddetli karaciğer yetmezliği (Child-Pugh C) olan hastalarda incelenmemiştir (bkz. Özel kullanım uyarıları ve önlemleri ve Pozoloji ve uygulama şekli).
Böbrek yetmezliği:
Bir klinik farmakoloji çalışmasında sorafenibin farmakokinetiği, böbrek fonksiyonları normal olan olgulara ve hafif (CrCl 50-80 mL/dk), orta dereceli (CrCl 30 ile < 50 mL/dk) ya da diyaliz gerektirmeyen şiddetli böbrek yetmezliği (CrCl < 30 mL/dk) olan olgulara tek doz 400 mg uygulamasından sonra değerlendirilmiştir. Sistemik sorafenibe maruz kalma ve renal fonksiyon arasında ilişki gözlenmemiştir. Hafif, orta dereceli ya da diyaliz gerektirmeyen şiddetli böbrek yetmezliği nedeniyle doz ayarlaması gerekli değildir (bkz. Pozoloji ve uygulama şekli).
Yaşlılar (65 yaş üzeri):
Demografik verilerin analizleri, yaşa göre doz ayarlaması yapılmasının gerekli olmadığını göstermektedir.
Pediyatrik hastalar:
5.3. Klinik öncesi güvenlilik verileri
Karsinojenez, Mutajenez, Fertilite bozukluğu
Sorafenibin klinik öncesi güvenlilik profili fareler, sıçanlar, köpekler ve tavşanlarda değerlendirilmiştir.
Tekrarlı doz toksisite çalışmalarında, çeşitli organlarda hafif ile orta dereceli değişmeler olduğu (dejenerasyonlar ve rejenerasyonlar) ortaya çıkarılmıştır.
Genç ve büyümekte olan köpeklere tekrarlı doz uygulamasından sonra, kemik ve
2
dişler üzerinde etkiler gözlenmiştir. Bu değişiklikler, günlük 600 mg/m2 vücut yüzey alanı sorafenib dozunda (vücut yüzey alanı temelinde önerilen klinik doz olan 500 mg/m2’nin 1.2 katına eşdeğer) femoral büyüme plağında düzensiz kalınlaşmalar, 200 mg/m /gün düzeyinde değişen büyüme plağına komşu kemik iliğinde hiposelülarite ve 600 mg/m2/gün düzeyinde dentin bileşiminde değişikliklerden oluşmaktaydı. Erişkin köpeklerde benzeri etkiler indüklenmemiştir.
Bir in vitro memeli hücresi çalışmasında (Çin hamsteri overleri), metabolik aktivasyon varlığında klastojenisite (kromozomal aberrasyonlar) için pozitif genotoksik etkiler elde edilmiştir. Üretim prosesi sırasında oluşan ve aynı zamanda bitmiş ilaç hammaddesinde de bulunan (< %0.15) bir ara madde, bir in vitro bakteriyel hücre incelemesinde (Ames testi) mutajenez yönüyle pozitiftir. Sorafenib, Ames testinde (%0.34 düzeyinde ara madde) ve bir in vivo fare mikronukleus incelemesinde genotoksik bulunmamıştır.
Sorafenib ile karsinogenesite çalışmaları yürütülmemi ştir.
Sorafenib ile hayvanlarda fertilite üzerindeki etkiyi değerlendirme amaçlı spesifik
çalışmalar yapılmamıştır. Ancak erkek ve dişi fertilitesi üzerinde bir advers etki
beklenebilir, çünkü hayvanlarda yürütülen tekrarlı doz çalışmaları, erkek ve dişi
üreme organlarında değişimler olduğunu göstermiştir. Tipik değişimler, sıçanların
testis, epididim, prostat ve seminal veziküllerinde dejenerasyon ve retardasyon
bulgularından oluşmaktadır ve bu etkiler günlük 150 mg/m vücut yüzey alanı
sorafenib dozunda açık bir şekilde belirmiştir (vücut yüzey alanı temelinde
önerilen 500 mg/m ’lik klinik dozun yaklaşık üçte biri). Dişi sıçanlarda korpus
luteumda santral nekroz ve overlerde foliküler gelişim duraklaması görülmüştür
ve gözlenen en düşük etki düzeyi 30 mg/m2/gün olmuştur. Köpeklerde 600 22 mg/m /gün dozunda testislerde tübüler dejenerasyon ve 1200 mg/m /gün dozunda
oligospermi görülmüştür.
Sorafenibin sıçanlar ve tavşanlara uygulandığında embriyotoksik ve teratojen olduğu gösterilmiştir. Gözlenen etkiler maternal ve fetal vücut ağırlığında azalma, fetal rezorpsiyon sayısında artış ve eksternal ve viseral malformasyon sayısında artıştan oluşmaktadır. Sıçanlarda oral 6 mg/m /gün dozunda ve tavşanlarda 36 mg/m2/gün dozunda advers fetal sonlanımlar gözlenmiştir (bkz. Özel kullanım uyarıları ve önlemleri ve Gebelik ve laktasyon).
6. FARMASÖTİK ÖZELLİKLER
6.1. Yardımcı maddelerin listesi
- Kroskarmeloz sodyum
- Mikrokristalimsi selüloz
- Hidroksipropilmetil selüloz
- Sodyum lauril sülfat
- Magnezyum stearat
- Makrogol
- Titanyum dioksit
6.2. Geçimsizlikler
Bilinen bir geçimsizliği bulunmamaktadır.
6.3. Raf ömrü
6.4. Saklamaya yönelik özel tedbirler
6.5. Ambalajın niteliği ve içeriği
6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemler
Kullanılmamış olan ürünler ya da atık materyaller, "Tıbbi Atıkların Kontrolü Yönetmeliği" ve "Ambalaj ve Ambalaj Atıklarının Kontrolü Yönetmeliği"ne uygun olarak imha edilmelidir.
 HIV ve Aids
HIV, Human Immunodeficiency Virus’dür (İnsanlarda Bağışıklık Sistemini Bozan
Virüsdür). Bu virüs AIDS hastalığına sebep olur.
HIV ve Aids
HIV, Human Immunodeficiency Virus’dür (İnsanlarda Bağışıklık Sistemini Bozan
Virüsdür). Bu virüs AIDS hastalığına sebep olur. |
 İnme
İnme, beynin hasar görmesinin sonucudur. Bu hasar, beynin bir kısmındaki ya bir kanama
ya da akut kan eksikliği nedeniyle o kısmın geçici ya da kalıcı olarak işlevini yapamamasına
yol açar.
İnme
İnme, beynin hasar görmesinin sonucudur. Bu hasar, beynin bir kısmındaki ya bir kanama
ya da akut kan eksikliği nedeniyle o kısmın geçici ya da kalıcı olarak işlevini yapamamasına
yol açar. |
İLAÇ EŞDEĞERLERİ
| Eşdeğer İlaç Adı | Barkodu | İlaç Fiyatı |
|---|---|---|
| PROKINIB | 8699702099027 | 42,033.80TL |
| SOFEXAN | 8699540033658 | 42,033.80TL |
| Diğer Eşdeğer İlaçlar |
 |
Şizofrenlik Şizofrenliğin psikiatrik teşhisi hakkında çok fazla anlaşmazlık vardır. Bu sayfadaki bilgiler, şizofrenliğin teşhisi, nedenleri ve tedavisi hakkındaki faklı teoriler hakkında bilgi verecektir. |
 |
Ruh ve Akıl Sağlığımızı Geliştirmek İyi akıl ve ruh sağlığı sahip olmaktan ziyade, yaptığınız şeylerdir. Akıl ve ruhsal olarak sağlıklı olmak için kendinize değer vermeli ve kendinizi kabul etmelisiniz. |
 |
Artrit Artrit, oldukça yaygın bir hastalıktır ancak iyi anlaşılamamıştır. Aslında “artrit” tek bir hastalığın adı değildir; eklem ağrısı veya eklem hastalıklarını adlandırmanın gayri resmi yoludur. |
İLAÇ GENEL BİLGİLERİ
Bayer Türk Kimya San. Tic. Ltd. Şti.
| Geri Ödeme Kodu | A11789 |
| Satış Fiyatı | 42033.8 TL [ 9 Jun 2026 ] |
| Önceki Satış Fiyatı | 42033.8 TL [ 22 May 2026 ] |
| Original / Jenerik | Original İlaç |
| Reçete Durumu | Normal Reçeteli bir ilaçdır. |
| Barkodu | 8699546090044 |
| Etkin Madde | Sorafenib |
| ATC Kodu | L01EX02 |
| Birim Miktar | 200 |
| Birim Cinsi | MG |
| Ambalaj Miktarı | 112 |
| Antineoplastik ve İmmünomodülatör Ajanlar |
| İthal ( ref. ülke : Yunanistan ) ve Beşeri bir ilaçdır. |