KALETRA 200 mg/50 mg 120 film tablet {Abbott} Kısa Ürün Bilgisi
{ Lopinavir + Ritonavir }
1. BEŞERİ TIBBİ ÜRÜNÜN ADI
KALETRA® (200 mg/50 mg) film tablet2. KALİTATİF VE KANTİTATİF BİLEŞİM
Her bir film tablet, farmakokinetik güçlendirici olarak 50 mg ritonavir ile birlikte formüle edilmiş olan 200 mg lopinavir içerir.
Yardımcı maddeler için 6.1.’e bakınız.
3. FARMASÖTİK FORMU
Film Tablet
Sarı renklidir. ’Abbott’ logosu ve "KA" harfleri basılıdır.
4. KLİNİK ÖZELLİKLER 1 Terapötik endikasy onlar
KALETRA®, diğer antiretroviral ajanlarla kombine olarak erişkinlerde ve 2 yaşın üzerindeki çocuklarda HIV-1 enfeksiyonu tedavisi için endikedir.
KALETRA® ile en çok ürünün antiretroviral tedavinin başındaki hastalarda kullanımından tecrübe elde edilmiştir. Daha önce ağır proteaz inhibitör tedavisi görmüş hastalardaki veriler sınırlıdır. KALETRA® tedavisinde başarısız olunmuş hastalardaki kurtarma tedavisi verileri sınırlıdır.
HIV-1 enfeksiyonlu proteaz inhibitör tedavisi geçirmiş hastalarda KALETRA® seçimi, bireysel viral direnç testine ve hastanın tedavi geçmişine göre belirlenmelidir. (bakınız bölüm 44. ve 5.1)
4.2. Pozoloji ve uygulama şekli
Pozoloji/uygulama sıklığı ve süresi:
KALETRA®, HIV enfeksiyonu tedavisinde deneyimli doktorlar tarafından reçete edilmelidir.
Yetişkinler ve ergenler:
Kaletra Tabletler, bütün olarak yutulmalı ve çiğnenmemeli, kırılmamalı ya da ezilmemelidir. Yutma güçlüğü olan hastalar için oral solüsyon mevcuttur.
Önerilen oral doz aşağıdaki gibidir: Daha evvel tedavi görmemiş hastalarda:
• Kaletra Tabletler 400/100 mg (2 Tablet) günde iki kez yemeklerle ya da tek başına
• Kaletra Tabletler 800/200 mg (4 Tablet) günde bir kez yemeklerle ya da tek başına
Daha evvel tedavi görmüş hastalarda:
• Kaletra Tabletler 400/100 mg (2 Tablet) günde iki kez yemeklerle ya da tek başına Günde bir kez Kaletra uygulaması daha evvel tedavi görmüş hastalarda çalışılmamıştır.
Özel Popülasyonlara ilişkin Ek Bilgiler Karaciğer yetmezliği
4.3. Kontrendikasyonlar
Böbrek yetmezliği
Böbrek yetmezliği olan hastalarda doz ayarlaması gerekli değildir. KALETRA® şiddetli böbrek yetmezliği olan hastalarda kullanıldığında dikkatli olunması gerekir (bakınız bölüm 4.4).
Pediyatrik popülasyon (2 yaş ve üstü)
Pediyatrik hastalarda günde bir kez alınan lopinavir/ritonavir değerlendirilmemiştir. KALETRA® efavirenz, nevirapin veya amprenavir ile eş zamanlı alınmayan yetişkin dozu (günde iki defa 400/100 mg), 35 kg veya üstündeki çocuklarda veya Vücut Yüzey Alanı (BSA)* 1.4 m2 veya daha büyük olan çocuklarda kullanılabilir. Vücut ağırlığı 35 kg veya BSA 0.5 ila 1.4 m2 arasında olan ve tablet yutabilen çocuklar için lütfen aşağıdaki doz tablosuna bakınız. KALETRA® Oral Solüsyon BSA 0.5 m2 den küçük veya güvenle tablet yutamayan çocuklar için uygundur.
*Vücut yüzey alanı aşağıdaki denklemden hesaplanabilir:
BSA (m2) = V [Boy (cm) x Ağırlık (kg)] / 3600
Aşağıdaki tablo KALETRA® 100/25 mg tabletlerin BSA’ya dayanan doz talimatlarını içermektedir:
Efavirenz, nevirapin veya amprenavir ile eş zamanlı alınmayan ve BSA’ya dayanan pediyatrik doz talimatı | |
2 Vücut Yüzey Alanı (m ) | Günde iki kere tavsiye edilen 100/25 mg tablet sayısı |
> 0.5 ila < 0.9 | 2 tablet (200/50 mg) |
> 0.9 ila < 1.4 | 3 tablet (300/75 mg) |
> 1.4 | 4 tablet (400/100 mg) |
Eş zamanlı tedavi: Efavirenz, nevirapin veya amprenavir
Aşağıdaki tablo çocuklarda efavirenz, nevirapin veya amprenavir ile eş zamanlı alınan ve BSA’ya dayanan 100/25 mg tabletlerin doz talimatını içermektedir:
Efavirenz, nevirapin veya amprenavir ile eş zamanlı alınan ve BSA’ya dayanan pediyatrik doz talimatı | |
0.5 ila < 0.8 | 2 tablet (200/50 mg) |
> 0.8 ila < 1.2 | 3 tablet (300/75 mg) |
> 1.2 | 4 tablet (400/100 mg) |
2 yaşın altındaki çocuklarda
Güvenlik ve etkinlik verilerinin yetersiz olması nedeniyle 2 yaşın altındaki çocuklarda KALETRA® kullanımı tavsiye edilmemektedir.
Geriatrik popülasyon
Genel olarak, sıklıkla düşük karaciğer, böbrek veya kalp fonksiyonları gösteren ve beraberinde hastalığı olan veya diğer ilaç tedavileri gören yaşlı hastaların ilaç alımında ve lopinavir/ritonavirin izlenmesinde gerekli tedbirler alınmalıdır.
4.3. Kontrendikasyonlar
Liponavir/ritonavir liponavir, ritonavir veya yardımcı maddelerden birine aşırı duyarlılığı olduğu bilinen hastalarda kontrendikedir.
KALETRA® her ikisi de CYP3A izoformu olan P450’ nin inhibitörleri olan lopinavir ve ritonavir içerir. KALETRA® CYP3A’ ya yüksek oranda bağımlı ilaçlarla klerens açısından ve kullanıldığında ciddi ve/veya yaşamı tehdit edebilen yüksek plazma konsantrasyonları yaratan ilaçlarla birlikte verilmemelidir. Bu ilaçlar astemizol, terfenadin, oral midozolam ( parenteral alınan midozolam uyarısı için bölüm 4.5’e bakınız), triazolam, sisaprid, pimozid, amiodaron, ergot alkaloidleri ( ergotamin, dihidroergotamin, ergonovin ve metilergonovin gibi) ve vardenafil.
St. John’s Wort (Hypericum perforatum) içeren bitkisel preparatlar, plazma konsantrasyonlarında düşme riskine ve lopinavir ile ritonavirin klinik etkisinin azalmasına sebebiyet vermesine bağlı olarak lopinavir ve ritonavir alınırken kullanılmamalıdır. (bakınız bölüm 4.5).
4.4. Özel kullanım uyarıları ve önlemleri
Dikkat!
KALETRA® Film Tablet, daha önceden kullandığınız KALETRA® Yumuşak Kapsül ile karıştırılmamalıdır. KALETRA® Film Tablet kullanmaya başladıktan sonra aynı ilacı kullanmaya devam ediniz. KALETRA® Film Tablet bir seferde 2 tablet birlikte olmak üzere, 12 saatte bir 2 tablet şeklinde kullanılır. Başladığınız ürünle tedaviye devam ediniz. Detaylı bilgi için doktorunuza veya eczacınıza danışınız.
UYARILAR Antimikobakteriyel
Standart dozdaki lopinavir/ritonavir, rifampisin ile birlikte uygulanmamalıdır çünkü lopinavir konsantrasyonlarındaki büyük düşüşler, terapötik etkide anlamlı ölçüde azalmaya neden olabilmektedir.
Kortikosteroidler
Lopinavir/ritonavir ve flutikazon propionatın birlikte uygulanması sonucunda flutikazon propionatın plazma düzeyleri anlamlı ölçüde artar ve serum kortizol konsantrasyonları azalır. Ritonavir, inhalasyon yoluyla veya intranazal olarak uygulanan flutikazon propionatla birlikte uygulandığında Cushing sendromu ve adrenal süpresyon dahil sistemik kortikosteroid etkiler bildirilmiştir. Budesonid gibi flutikazona benzer biçimde metabolize edilen diğer inhale kortikosteroidlerle lopinavir/ritonavirin birlikte uygulanması ile rastlanılan benzer bulgular göz ardı edilemez. Bu inhale ya da intranazal olarak uygulanan kortikosteroidler, lopinavir/ritonavir ile birlikte uygulandığında özel dikkat gösterilmelidir.
Erektil fonksiyon bozukluğu ilaçları
Lopinavir/ritonavir alan hastalara sildenafil, tadalafil ya da vardenafil reçetelenirken özellikle dikkatli olunmalıdır. Lopinavir/ritonavir ile bu ilaçların birlikte uygulanmasının bu ilaçların konsantrasyonlarını önemli ölçüde artırması beklenir ve hipotansiyon, ve ereksiyonda uzama dahil sildenafil ile bağlantılı advers olaylarda bir artışla sonuçlanabilir (bakınız bölüm 4.5).
Bitkisel ilaç etkileşimleri
St. John’s Wort (Hypericum perforatum) içeren bitkisel ürünler, proteaz inhibitörlerinin plazma konsantrasyonlarını azaltabileceğinden lopinavir/ritonavir alan hastalar tarafından kullanılmamalıdır. Bu durum lopinavire ya da proteaz inhibitörlerinin terapötik sınıfına direnç gelişimi ve terapötik etkinin kaybı ile sonuçlanabilir (bakınız Bölüm 4.5).
HMG-CoA redüktaz inhibitörleri
Lopinavir/ritonavir ile simvastatin veya lovastatinin bir arada kullanılması önerilmemektedir. Rabdomiyoliz dahil miyopati gibi ciddi reaksiyonlar için potansiyeli artırabileceğinden lopinavir/ritonavir dahil HIV proteaz inhibitörleri, CYP3A4 yolu (örn. atorvastatin) vasıtasıyla metabolize edilen rosuvastatin ya da diğer HMG-CoA redüktaz enzimleri ile eşzamanlı kullanılırsa dikkat edilmelidir (bakınız bölüm 4.5).
Tipranavir: Çoklu tedavi görmüş olan HIV - pozitif hastalarda ikili proteaz inhibitör kombinasyon tedavisi uygulanan bir klinik çalışmada, günde iki kere 500 mg tipranavir ile ritonavir (günde iki kez 200 mg) ve günde iki kere lopinavir/ritonavir (400/100 mg)’ın birlikte uygulanması, lopinavir EAA (Eğri Altında kalan Alan) ve Cmin değerlerinde sırasıyla %47 ve %70 azalmayla sonuçlanmıştır. KALETRA® ve düşük doz ritonavirle birlikte tipranavir uygulanması bu nedenle önerilmez.
Hiperglisemi
Pazarlama sonrası gözetimlerde proteaz inhibitörleri alan HIV enfeksiyonlu hastalarda yeni başlayan diabetes mellitus, hiperglisemide veya mevcut diabetes mellitusta alevlenme bildirilmiştir. Bu olayların tedavisinde bazı hastalarda ya insüline başlanması ya da dozunun ayarlanması ya da oral hipoglisemik ajanlar gerekmiştir. Bu olguların bazılarında diyabetik ketoasidoz gelişmiştir. Proteaz inhibitörü tedavisini bırakan bu hastalardan bazı vakalarda hiperglisemi kalıcılık göstermiştir. Bu vakalar klinik pratik esnasında gönüllü olarak bildirildiğinden sıklık tahminleri yapılamaz ve proteaz inhibitörü tedavisi ile bu vakalar arasındaki raslantısal ilişki kanıtlanamamıştır.
Pankreatit
KALETRA® alan hastalarda belirgin trigliserid yükselmeleri gelişenler dahil pankreatit olguları bildirilmiştir. Bazı vakalarda ölüm gözlemlenmiştir. Lopinavir/ritonavir ile raslantısal bir ilişkisi kanıtlanmamış olmasına rağmen belirgin trigliserid yükselmesi pankreatit gelişim için bir risk faktörüdür (bkz. 4.4. Lipid yükselmeleri). Lopinavir/ritonavir tedavisi esnasında ileri HIV hastalığı olan hastalarda trigliserid yükselmesi ve pankreatit ve hikayesinde pankreatit olan hastalarda pankreatitin yeniden nüks etmesi riski olabilir .
KALETRA® HIV enfeksiyonu veya AIDS için çare değildir. HIV enfeksiyonunun cinsel yolla veya kan temasıyla başkalarına bulaşma riskini azaltmaz. Gerekli önlemler alınmalıdır. KALETRA® kullanan kişiler hala enfeksiyon geliştirebilir veya HIV hastalığı ve AIDS ile ilgili diğer rahatsızlıkları geçirebilirler.
ÖNLEMLER
Karaciğer yetmezliği:
KALETRA®, başlıca olarak karaciğerde metabolize olur. Bu nedenle karaciğer fonksiyon bozukluğu bulunan hastalara uygulandığında dikkat edilmelidir. KALETRA®, şiddetli karaciğer yetmezliğinde çalışılmamıştır. Farmakokinetik veriler orta karaciğer yetmezliği bulunan HIV ve HCV’nin ikisi ile de enfekte olmuş hastalarda lopinavir plazma konsantrasyonlarının yaklaşık %30 artış bunun yanısıra plazma proteinlerine bağlanmada düşüşler olduğunu ileri sürer (bkz. Bölüm 5.2). Altta yatan bir hepatit B ya da C hastalığı ya da tedaviden önce transaminazlarında belirgin ölçüde artışlar görülen hastalar, daha ileri safhada transaminaz yükselmeleri gelişimi bakımından artmış bir risk taşıyabilirler. Bazı ölümler dahil karaciğer fonksiyon bozukluğuna dair pazarlama sonrası bildirimler bulunmaktadır. Bu durum genellikle altta yatan kronik hepatit ya da sirozun tedavisi için çoklu tedavi almakta olan ileri düzeyde HIV hastalığına sahip hastalarda meydana gelmiştir. KALETRA® ile bu vakalar arasındaki raslantısal bir ilişki kanıtlanamamıştır. Bu hastalarda özellikle KALETRA® tedavisinin ilk birkaç ayı boyunca artmış AST/ALT’nin izlenmesi düşünülmelidir.
Direnç/Çapraz direnç:
Proteaz inhibitörleri arasında çeşitli ölçülerde çapraz direnç gözlemlenmiştir. Proteaz inhibitörlerinin ardışık uygulanmasının etkililiğinde KALETRA® tedavisinin etkisi araştırma altındadır (bkz. Mikrobiyoloji)
Hemofili:
Proteaz inhibitörleri ile tedavi edilen tip A ve B hemofili hastalarında spontan deri hematomları ve hemartroz dahil kanamalarda artış olduğu bildirilmiştir. Bazı hastalara ilave faktör VIII verilmiştir. Bildirilen olguların yarısından fazlasında proteaz inhibitörleriyle tedaviye devam edilmiş veya tedavi kesilmişse yeniden başlanmıştır. Proteaz inhibörleri ve bu vakalar arasında ne raslantısal bir ilişki ne de bir etki mekanizması kanıtlanmıştır.
Yağ redistribüsyonu ve metabolik bozukluklar
Antiretroviral tedavi alan hastalarda genel obezite, dorsoservikal bölgede yağ toplanması (buffalo hump), yüzde incelme (facial wasting), göğüs büyümesi ve cushingoid görünüm gözlemlenmiştir. Bu vakaların mekanizması ve uzun dönemdeki neticeleri şu an bilinmemektedir. Bir sebep sonuç ilişkisi kanıtlanmamıştır.
Lipid yükselmeleri
KALETRA® tedavisi, total kolesterol ve trigliserid konsantrasyonunda artışlarla sonuçlanır. KALETRA® tedavisine başlamadan önce ve tedavi boyunca düzenli aralıklarla kolesterol ve trigliserid testleri yapılmalıdır. Yağ düzensizlikleri klinik açıdan uygun olacak şekilde tedavi edilmelidir.
immün Reaktivasyon Sendromu
Lopinovir/ritonavir dahil olmak üzere kombinasyon antiretroviral tedavi ile tedavi olan HIV infeksiyonlu hastalarda immün rekonstitüsyon sendromu bildirilmiştir. Kombinasyon antiretroviral tedavinin başlangıç aşamasında bağışıklık sistemi yanıt verdiğinde hastalarda, ileri düzeyde değerlendirmeyi ve tedaviyi gerektiren, asemptomatik veya rezidüel fırsatçı enfeksiyonlara (mycobacterium avium enfeksiyonu, sitomegalovirüs, pnömokistik carini pnömonisi ya da tüberkülozis gibi) yanıt olarak bir inflamatuar yanıt gelişebilir.
Osteonekroz
Etiolojinin çok faktörlü olduğunun düşünülmesine rağmen (kortikostreoid kullanımı, alkol tüketimi, ciddi immunosüpresyon, yüksek vücut kütle indeksi içeren), özellikle ileri derece HIV-hastalığı taşıyan veya uzun süre antiretroviral tedaviye (CART) maruz kalan hastalarda osteonekroz vakaları rapor edilmiştir. Hastalara eklem ağrısı, tutukluk ve hareket etmede zorluk şikayetleri varsa medikal yardım almaları tavsiye edilmelidir.
PR aralığında uzama ve ürünle indüklenmiş QT aralığında uzama
Lopinavir/ritonavirin bazı hastaların PR aralığında hafif ve semptomatik olmayan uzamaya neden olduğu gösterilmiştir. Lopinavir/ritonavir alan, altında yapısal kalp hastalığı yatan ve önceden var olan iletim sistemi anormallikleri olan hastalarda veya PR aralığını uzattığı bilinen ilaçlar (verapamil veya atazanavir gibi) alan hastalarda seyrek 2. veya 3. derece atriyoventriküler blok bildirilmiştir. KALETRA bu gibi hastalarda dikkatle kullanılmalıdır. (bakınız bölüm 5.1)
4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleri
4.3. Kontrendikasyonlar
KALETRA®, CYP3A tarafından metabolize edilir. KALETRA® ve CYP3A’yı indükleyen ilaçların birlite uygulanması, lopinavir plazma konsantrasyınlarını düşürür ve terapötik etkisini azaltır (bkz. Bölüm 4.4.). Her ne kadar ketokonazolün bildirilmemesine rağmen CYP3A’yı inhibe eden diğer ilaçların KALETRA® (lopinovir/ritonavir) ile birlikte uygulanması lopinavir plazma konsantrasyonlarını artırır.
Anti-HIV ajanlar
- Nükleozid Ters Transkriptaz İnhibitörleri (NRTI’ler)
Stavudin ve Lamivudin: KALETRA® klinik çalışmalarda tek başına ya da stavudin ve lamivudin ile kombine olarak verildiğinde lopinavirin farmakokinetiklerinde bir değişim gözlenmemiştir.
Didanosin: Didanosinin boş mideyle alınması önerilmektedir; bu nedenle didanosin KALETRA® Tabletleriyle birlikte tok karnına verilebilir.
Zidovudin ve Abakavir: KALETRA® glukuronidasyonu indüklemektedir, bu nedenle KALETRA®, zidovudin ve abacavirin plazma konsantrasyonlarını azaltma potansiyeline sahiptir. Bu potansiyel etkileşimin klinik anlamı bilinmemektedir.
Tenofovir: Bir çalışma, KALETRA® nın tenofovir konsantrasyonlarını artırdığını göstermiştir. Bu etkileşimin mekanizması bilinmemektedir. KALETRA® ve tenofovir kullanmakta olan hastalar tenofovire bağlı advers olaylar bakımından izlenmelidir.
Tümü: Proteaz inhibitörleri (PIs) özellikle nükleozid olmayan ters transkriptaz inhibitörleri (NRTIs) ile kombine kullanıldığında artmış CPK, miyalji ve nadiren rabdomiyoliz bildirilmiştir.
- Nükleozid Olmayan Ters Transkriptaz İnhibitörleri (NNRTI’ler)
Nevirapin:
Sağlıklı gönüllülerde KALETRA® ile nevirapinin birlikte uygulanması esnasında lopinavirin farmakokinetiğinde hiçbir değişiklik görülmemiştir. HIV-pozitif pediyatrik deneklerdeki bir çalışmanın sonuçları, nevirapin ile birlikte uygulanması esnasında lopinavir derişimlerinde bir düşüş olduğunu açığa çıkarmıştır. HIV-pozitif yetişkinlerde nevirapinin etkisi, pediyatrik deneklerinkine benzerdir ve lopinovir derişimleri düşebilir. Bu farmakokinetik etkileşimin klinik anlamı bilinmemektedir. Günde bir kez KALETRA® uygulaması, nevirapin ile birlikte alınmamalıdır.
Efavirenz
KALETRA® nın günde bir kez alınan 600 mg efavirenz ile birlikte alındığında uygulan dozu günde iki kez alınan 500/125 mg’a yükseltildiğinde, lopinavir konsantrasyonları günde tek başına iki kez alınan 400/100 mg KALETRA® nınkine aşağı yukarı yakındır. Açığa çıkan lopinavir, tek başına günde iki kere alınan 400/100 mg KALETRA® nınki ile karşılaştırıldığında %6 ila %12 oranında artmıştır.
Not: Efavirenz ve nevirapin CYP3A aktivitesini indükler ve bu nedenle KALETRA® ile birlikte kullanıldıklarında diğer inhibitörlerin plazma konsantrasyonlarını düşürme potansiyeline sahiptirler. Günde bir kez KALETRA® uygulaması, efavirenz ile birlikte alınmamalıdır.
Delavirdin:
Delavirdin, lopinavir plazma konsantrasyonlarını artırma potansiyeline sahiptir.
- Diğer HIV proteaz inhibitörleriyle (PI’ler) birlikte uygulama:
KALETRA® (günde iki defa 400/100 mg) kararlı durumda kontrollü sağlıklı gönüllülerde yapılan çalışmalarda ritonavir olmadan her bir HIV proteaz inhibitörünün klinik dozlarına relatif olarak azaltılmış amprenavir, indinavir, nelfinavir ve sakinavir ile kombine edilerek araştırılmıştır. Ritonavirle güçlendirilmiş amprenavir ve sakinavir rejimlerine ilişkin yayınlanmış farmakokinetik verilere de karşılaştırmalar yapılmıştır. Ekm olarak, ilave ritonavir lopinavir farmakokinetiği üzerindeki etkisi tartışılmaktadır. Ritonavirle güçlenidirilmiş proteaz inhibitör rejimlerin eski karşılaştırmalarının dikkatle yorumlanması gerektiğine dikkat edin (bakınız aşağıda kombinasyonların ayrıntıları).
Güvenlilik ve etkinlik yönünden Klatra ile kombine edilecek uygun HIV proteaz inhibitörü dozları bilnmemektedir. Bu nedenle KALETRA® nın PI’lerle konkomitan olarak uygulanması yakından izlenmelidir.
Amprenavir
4.2. Pozoloji ve uygulama şekli
). Günde bir kez alınan KALETRA® uygulaması amprenavir ile birlikte alınmamalıdır.
Fosamprenavir
Bir çalışma, KALETRA® ile fosamprenavirin birlikte uygulanmasının amprenavir ve lopinavir konsantrasyonlarını azalttığını göstermiştir. KALETRA® ve fosamprenavirin uygun dozlarının kombinasyonu güvenlilik ve etkililik bakımından kanıtlanmamıştır.
Indinavir
KALETRA® nın indinavir (günde 2 kez 600 mg indinavir + KALETRA®, tek başına günde 3 kez 800 mg indinavir nazaran benzer EAA, azalmış Cmaks ve artmış Cmin yaratır) konsantrasyonlarını artıması beklenir. Günde 2 kez 400 mg/100 mg KALETRA® ile birlikte uygulanan indinavir dozunun azaltılması gerekebilir.
Nelfinavir
4.2. Pozoloji ve uygulama şekli
). KALETRA®, nelfinavir ile birlikte kombinasyon tedavisinde günde bir kez olarak alınmamalıdır.
Ritonavir
Tekbaşına günde iki kez 400/100 mg KALETRA® (üç yumuşak jelatin kapsül) ile karşılaştırıldığı üzere KALETRA® ilave olarak 100 mg ritonavir (günde 2 kez) ile birlikte uygulandığında lopinavir EAA %33 ve Cmin %64 artar.
Sakinavir
KALETRA® nın sakinavir konsantrasyonlarını (günde 2 kez 800 mg sakinavir + KALETRA®, tek başına günde 3 kez 1200 mg sakinavire nazaran artmış EAA, artmış Cmaks ve artmış Cmin yaratır) artırması beklenir. Günde 2 kez 400 mg/100 mg KALETRA® ile birlikte uygulanan sakinavir dozunun azaltılması gerekebilir. Günde bir kez KALETRA® uygulaması, sakinavir ile birlikte çalışılmamıştır.
Diğer
Omeprazol ve ranitidin:
Sağlıklı gönüllülerle yapılan bir çalışmada günde iki kez KALETRA® 400/100 mg omeprazol ile veya ranitidin ile birlikte alındığında klinik olarak ilişkili herhangi bir etkileşim gözlenmemiştir. KALETRA® tabletleri asit düşürücü ajanlarla (omeprazol ve ranitidin) kombinasyonda doz ayarlamasına gerek olmadan kullanılabilir.
Antiaritmikler
: (amiodaron, bepridil, sistemik lidokain ve kinidin): KALETRA® ile birlikte uygulandığında konsantrasyonları artabilir. Dikkatli olunması ve mümkünse terapötik konsantrasyonun izlenmesi önerilir.
Antikanser Ajanları (örn. vinkristin, vinblastin): KALETRA® ile birlikte uygulandıklarında bu antikanser ajanlar ile bağdaştırılan advers etkilerin artmış potansiyeline neden olan serum konsantrasyonlarına sahip olurlar.
Antidepresanlar:
Bupropion: Bupropionun lopinavir/ritonavir ile eş zamanlı kullanımı hem bupropionun plazma seviyesini hem de aktif metabolitinin (hidroksibupropion) seviyesini düşürür.
Antikonvülsanlar: (fenobarbital, fenitoin, karbamazepin): Bu ilaçların CYP3A4’ü indükledikleri bilinmektedir ve lopinavir konsantrasyonlarını düşürebilir. Buna ek olarak, fenitoinin KALETRA® ile birlikte uygulanması, kararlı durum fenitoin konsantrasyonlarında orta düzeyde artış ile sonuçlanmıştır. KALETRA® ile birlikte uygulandığında fenitoin düzeyleri izlenmelidir.
Antikoagülanlar
: Varfarin konsantrasyonları, KALETRA® nın birlikte uygulanmasından etkilenebilir. INR’nin (uluslararası normalize oran) izlenmesi önerilmektedir.
Trazodon: Ritonavir ve trazodonun bir arada kullanılması trazodon konsantrasyonlarını artırabilir. Bulantı, baş dönmesi, hipotansiyon ve senkop advers etkileri gözlemlenmiştir. Eğer trazodon, KALETRA® gibi bir CYP3A4 inhibitörü ile birlikte kullanılacaksa, bu kombinasyon dikkatle kullanılmalı ve trazodonun daha düşük bir dozunun kullanılması düşünülmelidir.
Digoksin: Bir literatür raporu, ritonavir ve digoksinin birlikte uygulanmasının digoksin düzeylerinin anlamlı ölçüde artması ile sonuçlandığını göstermiştir. KALETRA® ve digoksin birlikte uygulandığında serum digoksin düzeylerinin uygun bir şekilde izlenmesi ile dikkat edilmelidir.
Ketokonazol ve itrakonazolün, KALETRA® vasıtasıyla artan serum konsantrasyonları olabilir. Ketokonazol ve itrakonazolün yüksek dozları (200 mg/gün’den daha fazla) önerilmemektedir.
Vorikonazol: Bir çalışmada her 12 saatte bir 100 mg ritonavir ile birlikte uygulanan vorikonazolün kararlı durum EAA değerini %39’luk bir ortalama ile düşdüğü gösterilmiştir. Bu nedenle vorikonazol kullanımının hastaya yarar/risk değerlendirmesi ispat edilmeden KALETRA® ve vorikonazolün birlikte uygulanmasından kaçınılmalıdır
Anti-infektifler: Kaletra ile birlikte uygulandığında klaritromisin EAA’sında orta düzeyde artışlar beklenmektedir. Böbrek ve karaciğer yetmezliği bulunan hastalarda klaritromisin dozunun azaltılması düşünülmelidir.
Buprenorfin: lopinavir/ritonavir (günde iki kez 400/100 mg doz) ile birlikte buprenorfin (günde 16 mg doz) alınması klinik olarak önemli bir etkileşim göstermemiştir. KALETRA® buprenorfin ile birlikte doz ayarlanmasına gerek kalmadan alınabilir.
Antimikobakteriyaller:
Rifabutin: Rifabutin ve KALETRA® 10 gün birlikte uygulandıklarında, rifabutin (ana ilaç ve aktif 25-O-desasetil metaboliti) Cmaks ve EAA sırasıyla 3.5 ila 5.7 kat artış göstermiştir. Bu verilere dayanarak, KALETRA® ile birlikte verildiğinde rifabutin dozunun %75 oranında azaltılması (gün aşırı veya haftada 3 defa 150 mg) önerilmektedir. Rifabutin dozunun daha fazla azaltılması da gerekebilir.
Rifampisin: Rifampisinin standard dozdaki KALETRA® ile birlikte kullanımı, KALETRA®’ya, proteaz inhibitörlerine ya da diğer antiretroviral ajanlara karşı virolojik yanıtta ve olası dirençte kayba neden olabilir. Tek başına günde iki kez 400/100 mg KALETRA® dozu ile karşılaştırıldığında rifampisinin günde iki kez 800/200 mg KALETRA® dozu ile birlikte uygulanması ile lopinovirde %57’ye kadar ve günde iki kez 400/400 mg KALETRA® dozu ile birlikte uygulanması ise %7’ye kadar düşüşe yol açmıştır. Rifampisin ile birlikte uygulanan yüksek dozlardaki KALETRA® ile ALT ve AST yükselmeleri bildirilmiştir ve bu durum doz uygulaması sekansına bağlı olabilir. Birlikte kullanılmaları düşünülüyorsa KALETRA®, rifampisinin eklenmesinden 10 gün önce standart dozda başlanmalıdır. Bundan sonra KALETRA® dozu artırılmalıdır. Karaciğer işlevinin yakın takibi yapılmalıdır.
Antiparazitler:
KALETRA® birlikte uygulandığında atovakuonun terapötik konsantrasyonlarında artış muhtemeldir. Atovakuonun dozunda artış yapılması gerekebilir.
Kortikosteroidler:
Deksametazon: Deksametazon, CYP3A4’ü indükleyebilir ve lopinavir konsantrasyonlarını düşürebilir.
Flutikazon propionat: Flutikazon propionatla KALETRA®’nın birarada kullanımında lopinavir/ritonavir, flutikazon propionat konsantrasyonlarını artırabilir. Dikkatle kullanılmalıdırlar. Özellikle uzun süreli kullanım için flutikazon propionata alternatifler düşünülmelidir (bkz. Bölüm 4.4. Uyarılar, ’İlaç etkileşimleri’ alt başlığı)
Dihidropiridin kalsiyum kanal blokörleri
: (örn. felodipin, nifedipin, nikardipin): Serum konsantrasyonları KALETRA® ile artabilir.
Lipid düşürücü ajanlar
:
Büyük ölçüde CYP3A4 metabolizmasına bağımlı olan lovastatin ve simvastatin gibi HMG-CoA redüktaz inhibitörleri KALETRA® ile birlikte uygulandıklarında plazma konsantrasyonlarının belirgin şekilde artması beklenir. Artmış HMG-CoA redüktaz inhibitörleri rabdomiyoliz dahil miyopatiye neden olabileceğinden bu ilaçların KALETRA® ile kombinasyonu önerilmez. Atorvastatin, metabolizma açısından CYP3A’ya daha az bağımlıdır. Atorvastatin ile KALETRA® eşzamanlı olarak verildiğinde atorvastatin Cmaks ve EAA değerlerinde sırasıyla ortalama 4.7 kat ila 5.9 kat artış gözlemlenmiştir. KALETRA® ile birlikte kullanıldığında, mümkün olan en düşük atorvastatin dozları verilmelidir. Rosuvastatin CYP3A’ya bağımlı değildir. Ancak, KALETRA® ile birlikte alındığında rosuvastatinin Cmaks ve EAA değerlerinde sırasıyla ortalama 5 kat ila 2 kat artış gözlenmiştir.
4.4. Özel kullanım uyarıları ve önlemleri
, ’İlaç etkileşimleri’ alt başlığı).
Fosfodiesteraz inhibitörleri:
Tadanafil ve sildenafil gibi CYP3A4 metabolizmasına bağımlı fosfodiesteraz inhibitörleri, KALETRA® yı da içeren ritonavir tedavisi ile birlikte alınmasının EAA değerlerinde yaklaşık olarak sırasıyla 2 kat ve 11 katı artışla sonuçlanması beklenmektedir. Bu durumda, hipertansiyon, senkop, görsel değişiklikler ve geç ereksiyonu içeren advers etkilerle ilişkili PDE5 inhibitöründe artışa neden olabilir. KALETRA® kullanan hastalara sildenafil ve tadanafil reçetelendirilirken advers etkilerin izlenmesi için özel önlemler alınmalıdır. KALETRA® yı da içeren ritonavir tedavisi ile birlikte vardenafilin eş zamanlı kullanımı vardenafilin EAA değerinde 49 kat artış ile sonuçlanması beklenir. KALETRA® nın vardenafil ile birlikte kullanılması kontrendikedir.
Erektil fonksiyon bozukluğu ajanları
4.4. Özel kullanım uyarıları ve önlemleri
, ’İlaç etkileşimleri’ alt başlığı).
Tadalafil: Tadalafil dikkatle ve azaltılmış doz olan her 72 saatte bir 10 mg’ı geçmeyecek şekilde ve advers etkiler bakımından sıkı takiple kullanılmalıdır (bkz. Bölüm 4.4. Özel kullanım uyarıları ve önlemleri, ’İlaç etkileşimleri’ alt başlığı).
Vardenafil: Vardenafil dikkatle ve azaltılmış doz olan her 72 saatte bir 2,5 mg’ı geçmeyecek şekilde ve advers etkiler bakımından sıkı takiple kullanılmalıdır (bkz. Bölüm 4.4. Özel kullanım uyarıları ve önlemleri, ’İlaç etkileşimleri’ alt başlığı).
Siklosporin, sirolimus ve takrolimus: KALETRA® ile birlikte kullanıldığında kontrendikasyonları artabilir. Bu ürünlerin plazma seviyeleri kararlı hale gelene kadar daha sık terapötik konsantrasyon takibi tavsiye edilir.
Bitkisel ilaçlar
KALETRA® kullanmakta olan hastalar St.Johns Wort içeren ürünlerle birarada kullanmamalıdırlar. Zira bu kombinasyon KALETRA®’nın plazma konsantrasyonlarında azalmaya yol açması beklenebilir. Bu etkiler, CYP3A4’ün indüksiyonu yüzünden olabilir ve terapötik etki ve direnç gelişimi ile sonuçlanabilir (bkz. Bölüm 4.4. Özel kullanım uyarıları ve önlemleri, ’İlaç etkileşimleri’ alt başlığı).
Midazolam
: Midazolam yaygın olarak CYP3A4 ile metabolize olur. KALETRA® ile birlikte kullanımı benzodiazepin konsantrasyonunda büyük bir artışa neden olabilir. 14 sağlıklı gönüllüde yapılan bir fenotip kokteyl çalışması EEA’da 13 katı ve parenteral midazolamda 4 katı kadar artış göstermiştir. Bu sebeple KALETRA® oral yolla alınan midazolam ile birlikte kullanılmamalıdır. Bunun yanında parenteral yolla alınan midazolam ile birlikte kullanılırken de dikkatli olunmalıdır. Şayet KALETRA® parenteral yolla alınan midazolam ile birlikte kullanılacaksa, yoğun bakım ünitesinde veya yakın klinik izleme ve solunum depresyonunda ve/veya uzun süreli sedasyonda uygun medikal idare sağlayacak benzer şartlarda verilmelidir. Midazolam için doz ayarlaması, eğer midazolam bir kerede tek dozdan fazla alınırsa özellikle düşünülmelidir.
Tıbbi ürünlerle etkileşim:
Lopinavir/ritonavir, P450 izoformu CYP3A’nın bir inhibitörüdür. Lopinavir/ritonavir ve esas olarak CYP3A tarafından metabolize edilen ürünlerin birlikte uygulanması, diğer tıbbi ürünün plazma konsantrasyonlarına yükselterek bu ilacın terapötik ve advers etkilerinin artmasına ve uzamasına neden olabilir (bkz. Bölüm 4.5. ve 4.3.)
HMG CoA redüktaz inhibitörleri simvastatin ve lovastatin metabolizması CYP3A’ya yüksek oranda bağlı olduğundan, KALETRA®’nın simvastatin veya lovastatin ile konkomitan kullanımı, rabdomilayiz dahil miyopati riskindeki artış nedeniyle önerilmemektedir. KALETRA®, CYP3A4 ile daha az oranda metabolize edilen rosuvastatin ve atorvastatin ile eşzamanlı olarak kullanıldığında dikkatli olunmalı ve dozların azaltılması göz önüne alınmalıdır. Bir HMG CoA redüktaz inhibitörü endike olduğunda pravastatin veya fluvastatin önerilir (bakınız bölüm 4.5).
KALETRA®, klorfeniramin, kinidin, eritromisin ve klaritromisin gibi QT aralığında uzama yaptığı bilinen tıbbi ürünlerle reçetelendirildiğinde özel önlemler alınmalıdır. KALETRA® birlikte kullanıldığı tıbbi ürünlerin konsantrasyonunu arttırabilir ve bu durum bu ürünlerin kardiyak advers etkisinin artmasıyla ilişkilendirilebilir. Klinik öncesi çalışmalarda kardiyak olaylar rapor edilmiştir, bu sebeple Klatra’nın poatnsiyal kardiyak etkisi göz ardı edilmemelidir.
İmmünosupresanlar
Bu ilaçların (siklosporin, sirolimus ve takrolimus) konsantrasyonları, KALETRA® ile birlikte uygulandığında artabilir. Bu ürünlerin kan düzeyleri stabilize olana kadar tedavi konsantrasyonlarının daha sık izlenmesi önerilir.
Metadon: KALETRA®’nın metadonun plazma konsantrasyonlarını düşürdüğü gösterilmiştir. Metadonun plazma konsantrasyonlarının izlenmesi önerilir.
Oral yada flaster olarak uygulanan kontraseptifler Etinil östradiol düzeyleri azalabildiğinden, östrojen bazlı oral kontraseptifler ya da flaster formundaki kontraseptifler ve KALETRA® birlikte uygulandığında alternatif veya ilave kontraseptif önlemler kullanılmalıdır.
Klinik açıdan anlamlı ilaç etkileşimleri beklenmeyen ilaçlar
İlaç etkileşim çalışmaları, desipramin (CYP2D6 prob), omeprazol veya ranitidin ile klinik olarak anlamlı etkileşim göstermemektedir.
Bilinen metabolik profillere göre normal böbrek ve karaciğer fonksiyonlarına sahip hastalarda KALETRA® ve fluvastatin, dapson, trimetoprim/sulfametoksazol, azitromisin veya flukonazol arasında klinik açıdan anlamlı etkileşimler beklenmemektedir.
Karsinojenesite ve mutajenesite
Lopinavir/ritonavir ile farelerde yapılan uzun-dönemli karsinojenisite çalışmalarında karaciğer tümörlerinin genotoksik olmayan, mitojenik indüksiyon görülmüş olup bu durum genel olarak insanlardaki risk açısından küçük bir anlama sahiptir. Sıçanlardaki karsinojenisite çalışmalarında tümörojenik bulgular elde edilmemiştir. Lopinavir/ritonavirin Ames bakteriyel ters mutasyon tayini, fare lenfoması tayini ve insan lenfositlerinin kromozom aberasyon tayinleri dahil bir dizi in vitro tayinde mutajenik ya da klastojenik olduğu bulunmamıştır. Lopinavir/ritonavir, fare mikronükleus testi in vivo tayinde mutajenik ya da klastojenik bulunmamıştır.
4.6. Gebelik ve laktasyon
Genel tavsiye
Lopinavir/ritonavirin gebe kadınlarda kullanım emniyeti kanıtlanmamıştır. Gerekli olmadıkça gebelik döneminde kullanılmamalıdır. Hasta hamile kaldığında veya hamilelik kararı aldığında doktorunu bilgilendirmesi gerektiği konusunda uyarılmalıdır.
Gebelik Dönemi
Gebelik Kategorisi: C
KALETRA®’nın gebe kadınlarda kullanımına iliskin yeterli veri mevcut değildir. Bu nedenle KALETRA® gerekli olmadıkça gebelik döneminde kullanılmamalıdır.
Laktasyon Dönemi
Emzirilen küçük çocuklardaki ciddi advers reaksiyon ve HIV transmisyonu potansiyeli yüzünden anneler kesinlikle bebeklerini emzirmemelidir. Sıçanlardaki çalışmalar lopinavirin süte geçtiğini göstermiştir. Lopinavirin insan sütüne geçip geçmediği bilinmemektedir.
4.7 Araç ve makina kullanımı üzerindeki etkiler
Araç veya makina kullanma yeteneği üzerindeki etkilere ilişkin çalışmalar yapılmamıştır.
4.8. İstenmeyen etkiler
KALETRA® nın güvenliliği 442’sinin günde iki kez 400/100 mg aldığı 612 hastada, Faz II/III klinik çalışmasında araştırılmıştır. Bazı çalışmalarda, KALETRA® efavirenz veya nevirapin ile birlikte kullanılmıştır.
KALETRA® ile ilişkilendirilen en yaygın advers olay, genellikle hafif ila orta şiddetteki ishaldir. 48 haftadan fazla bir sürede advers etkilere bağlı olarak tedaviye devam edememe yüzdesi yeni hastalarda % 4.5 ve deneyimli hastalarda % 9’dur. KALETRA® alan hastalarda hipertrigliseridemi gelişimini de içeren pankreatit vakalarının rapor edildiğine dikkat edilmelidir. Bunun yanında, KALETRA® tedavisi sırasında nadiren PR aralığında artış bildirilmiştir. (bakınız bölüm 4.4: pankreatit ve lipid yükselmeleri)
Kısmen nükleozid transkriptaz inhibitörleri ile kombine proteaz inhibitörleri ile artan CPK ( kreatin fosfokinaz), miyalji, miyosit ve nadiren rabdomiyolizis rapor edilmiştir.
Kombine antiretroviral tedavi, periferal ve yüzün deri altı yağında kaybı, karın içi yağında artış ve iç organlarda yağlanma, göğüs hipertrofisi ve dorsoservikal yağ dağılımı (bufalo hörgücü) da görülen HIV hastalarında vücut yağ dağılımında bozukluk ile ilişkilendirilmiştir.
Kombine antiretroviral tedavi, hipertrigliseridemi, hiperkolesterolemi, insülin direnci, hiperglisemi ve hiperlaktatemi gibi metabolik anormalliklerle ilişkiliendirilmiştir.
Kombine antiretroviral tedaviye (CART) başlama aşamasında ciddi bağışıklık eksikliği olan HIV enfekte hastalarda semptomatik olmayan iltihabik bir reaksiyon veya kalıntısal oportünist enfeksiyon görülebilir. (bakınız bölüm 4.4)
Lopinovir/ritonavirin de kombinasyon tedavisinde kullanıldığı 48 (Çalışma: M98-863) ve 360 haftalık çalışmada (faz II/III) ani tedavi gerektiren %2 ve/veya üzerinde orta yada ciddi advers olaylar aşağıda listelenmiştir.
Yetişkinler
4.8. İstenmeyen etkiler
4.8. İstenmeyen etkiler
system organ sınıfına gore düzenlenmiştir. Hepsi sıklık sınıflandırması içinde, istenmeyen etkiler ciddiyet azalışına göre verilmiştir.
Çok yaygın (>1/10); yaygın (> 1/100 ila < 1/10); yaygın olmayan (> 1/1.000 ila < 1/100); seyrek (>1/10.000 ila < 1/1.000); çok seyrek (< 1/10.000), bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor)
Yetişkinlerde yapılan klinik çalışmalarda görülen istenmeyen etkiler
Enfeksiyonlar ve enfestasyonlar
Yaygın olmayan
Grip sendromu, furonküloz, gastroenterit, bakteriyel enfeksiyon, otitis media, farenjit, tükürük bezi iltihabı, sinüzit, viral enfeksiyon
(Kist ve polipler dahil olmak üzere) iyi huylu kötü huylu ve tanımlanamayan neoplazmalar
Yaygın olmayan
Deride benign neoplazma, neoplazma ve kist
Kan ve lenfatik sistemi hastalıkları
Yaygın olmayan
Anemi, lökopeni ve lenfadenopati
Bağışıklık sistemi hastalıkları
Yaygın olmayan Alerjik reaksiyonlar
Endokrin hastalıkları
Yaygın olmayan
Erkek hipogonadizm, Cushing sendromu, hipotiroidizm
Metabolik ve beslenme bozukluklar
Yaygın olmayan
Avitaminoz, dehidratasyon, ödem, diabetes mellitus, iştah artışı, laktik asidoz, obezite, anoreksi, hiperglisemi, hipokolesterolemi, kilo alımı
Psikiyatrik hastalıklar
Yaygın
Uykusuzluk Yaygın olmayan
Anormal düşler, ajitasyon, anksiyete, apati, depresyon, konfüzyon, diskinezi, emosyonel labilite, düşük libido, sinirlilik, anormal düşünceler
Sinir sistemi hastalıkları
Yaygın
Baş ağrısı, parestezi Yaygın olmayan
Baş dönmesi, amnezi, ataksi, serebral enfarktüs, konvülsiyon, diskinezi, ensefalopati, ekstrapiramidal sendrom, yüz felci, hipertoni, migren, nöropati, periferik nörit, somnolans, titreme, tat duyusu kaybı, tat duyusu bozukluğu,
Göz bozuklukları
Yaygın olmayan
Görme anormalliği, göz bozukluğu
Kulak ve iç kulak bozuklukları
Yaygın olmayan Çınlama ve vertigo
Kardiyak hastalıkları
Yaygın olmayan
Atriyal fibrilasyon, palpitasyon, miyokard enfarktüsü1
Vasküler hastalıkları
Yaygın olmayan
Postural hipotansiyon, tromboflebit, vaskülit, variköz ven, derin ven trombozu
Solunum, göğüs bozuklukları ve mediastinal hastalıkları
Yaygın olmayan
Dispne, rinit, öksürük artışı, astım ve akciğer ödemi
Gastrointestinal hastalıklar
Yaygın İshal
Yaygın olmayan
Bulantı, kusma, karın ağrısı, anormal feçes, dispepsi, mide gazı, gastrointestinal bozukluk Seyrek
Abdomen genişlemesi, konstipasyon, kabızlık, ağız kuruluğu, disfaji, enterokolit, eruktasyon, özofajit, fekal inkontinans, gastrit, gastroenterit, hemorajik kolit, ağızda ülserasyonlar, pankreatit1, stomatit, ülseratif stomatit, periodontit, sialoadenit
Hepatobiliyer hastalıkları
Yaygın olmayan
Kolanjit, kolesistit, hepatit, hepatomegali, sarılık, karaciğerde yağ birikimi, karaciğerde hassasiyet
Deri ve deri altı doku hastalıkları
Yaygın
Döküntü, lipodistrofi, akne Yaygın olmayan
Alopesi, deride kuruluk, egzama, eksfoliyatif dermatit, makülopapüler döküntü, tırnak bozuklukları, pruritis, sebore, deride renk değişimi, deri ülseri, akne, terleme, ciltte strialar
Kas iskelet ve bağ doku ve kemik hastalıkları
Yaygın
Artralji, artroz, sırt ağrısı, eklem bozukluğu, kemik nekrozu ve myasthenia
Böbrek ve üriner hastalıkları
Yaygın olmayan
Böbrek taşı, idrarda anormallik, nefrit, albüminüri, hiperkalsinüri, hiperürisemi
Üreme sistemi ve meme hastalıkları
Yaygın olmayan
Anormal ejakülasyon, meme büyemesi, jinekomasti, impotans
Genel bozukluklar ve uygulama bölgesine ilişkin hastalıkları
Yaygın Asteni, ağrı Yaygın olmayan
Göğüs ağrısı, substernal göğüs ağrısı, ödem, hipertrofi, bitkinlik, ağrı, periferal ödem, ilaç etkileşimi
Araştırmalar
Çok yaygın (Derece 3 veya 4)
Trigliseritte artış, toplam kolesterolde artış, GGT’de artış Yaygın (Derece 3 veya 4)
Glukozda artış, amilazda artış, SGOT/AST de artış, SGPT/ALT’de artış, karaciğer fonksiyon testlerinde anormallik Yaygın olmayan
Glukoz toleransında azalma, kilo alma, kilo kaybı, bilirubinde artış, hormon seviyelerinde değişiklik, laboratuvar testlerinde anormallik, ilaç düzeyinde artış
1 Bu olayın ölümcül sonucu vardır.
2 Bakınız bölüm 4.4: pankreatit ve lipidler
Pediyatrik hastalar
Ani tedavi gerektiren advers olaylar
KALETRA®, 6 ay ila 12 yaş arası 100 pediyatrik hastada çalışılmıştır. Bir klinik çalışma esnasındaki advers olay profili, yetişkinlerinkine benzerdi. Kombinasyonunda KALETRA®’nın da yer aldığı Çalışma 940’da 48 haftaya kadar tedavi edilen herhangi şiddetteki pediyatrik hastalarda ilaca bağlı olarak en yaygın bildirilen bildirilen advers olaylar tat bozukluğu, kusma ve ishaldir.
Pediyatrik hastalarda yapılan klinik çalışmalarda görülen istenmeyen etkiler
Enfeksiyonlar ve enfestasyonlar
Yaygın
Viral enfeksiyon
Sinir sistemi hastalıkları
Yaygın
Tat almada bozukluk
Gastrointestinal hastalıklar
Yaygın
Konstipasyon, kusma, pankreatit*
Hepatobiliyer hastalıkları
Yaygın Hepatomegali
Deri ve deri altı doku hastalıkları
Yaygın
Döküntü, ciltte kuruluk
Genel bozukluklar ve uygulama bölgesine ilişkin hastalıkları
Yaygın Ateş
Araştırmalar
Yaygın
(Derece 3 veya 4)
Aktive kısmi tromboplastin zamanında artış, hemoglobinde azalma, trombosit azalması, sodium artışı, potasyum artışı, kalsiyum artışı, bilirubin artışı, SGPT/ALT artışı, SGOT/AST artışı, toplam kolesterolde artış, amilazda artış, ürik asitte artış, sodyumda azalma, potasyumda azalma, kalsiyumda azalma, nötrofilde azalma
*bakınız bölüm 4.4 pankreatit ve lipidler
Pazarlama sonrası deneyim
KALETRA® tedavisi alan hastalarda hepatit bildirilmiştir. Stevens Johnson Sendromu ve eritema multiforme bildirilmiştir. Bradiaritmi bildirilmiştir.
4.9. Doz aşımı ve tedavisi
KALETRA®’nın akut aşırı dozuyla ilgili olarak insanlarda deneyim bugün için sınırlıdır.
Köpeklerde gözlemlenen advers klinik belirtiler salivasyon artışı, emezis ve diyare/anormal dışkılamadır. Farelerde, sıçanlarda veya köpeklerde gözlemlenen toksisite belirtileri aktivitede azalma, ataksi, emasiyasyon, dehidratasyon ve tremorlardır.
KALETRA® aşırı dozu için spesifik bir antidot yoktur. KALETRA® aşırı dozunun tedavisi vital belirtilerin izlenmesi ve hastanın klinik statüsünün gözlemi dahil genel destekleyici önlemlerden ibarettir. Endike ise, absorbe olmamış aktif maddenin emezis veya gastrik lavajla eliminasyonu sağlanır. Absorbe olmamış aktif maddeni uzaklaştırılması için aktif karbon da kullanılabilir. KALETRA® yüksek oranda proteine bağlandığından, diyalizin aktif maddenin anlamlı biçimde uzaklaştırılmasında yararlı olması olası değildir.
5. FARMAKOLOJİK ÖZELLİKLER
5.1. Farmakodinamik özellikler
Farmakoterapötik grup: sistemik kullanım için antiviral ATC kodu: J05AE06
Mikrobiyoloji
Etki mekanizması: Lopinavir HIV-1 ve HIV-2 proteazların bir inhibitörü olup, gag-pol poliproteinin klivajını engelleyerek olgunlaşmamış, enfeksiyöz olmayan virüs üretimine yol açar.
İn vitro antiviral aktivite: Lopinavirin laboratuardaki ve klinikteki HIV suşlarına karşı in vitro antiviral aktivitesi, sırasıyla, akut olarak enfekte edilmiş lenfoblastik hücre dizilerinde ve periferik kan lenfositlerinde değerlendirilmiştir. İnsan serumunun yokluğunda, lopinavirin beş farklı HIV-1 laboratuar suşuna karşı ortalama %50 etkili konsantrasyonu (EC50) 10 ila 27 nm (0.006 - 0.017 mcg/ml, 1 mcg/ml, 1.6 microM’a eşdeğerdir) ve birkaç HIV-1 klinik izolatına karşı (n, 6’ya eşdeğerdir) 4 ila 11 nm (0.003 - 0.007 mcg/ml) aralığında değişmektedir. Yüzde 50 insan serumu varlığında lopinavirin bu beş laboratuar suşuna karşı ortalama EC50’si, 7-11 katlık bir azalmayı göstererek 65 ila 289 nM (0.04 - 0.18) mcg/ml aralığında değişmektedir. Lopinavir ve diğer proteaz inhibitörlerinin ya da ters transkriptaz inhibitörlerinin ilaç kombinasyonu aktivite çalışmaları henüz tamamlanmamıştır.
Direnç
Lopinavire karşı duyarlılığı azalmış olan HIV-1 izolatları in vitro olarak seçilmiştir.
Ritonavirin varlığı, lopinavire dirençli virüslerin in-vitro olarak seçilmesini etkilemediği gözükmektedir.
Çapraz direnç
Klinik öncesi çalışmalar
Proteaz inhibitörleri arasında değişen derecelerde çapraz direnç gözlemlenmiştir. Daha evvel tek bir proteaz inhibitörü ile tedavi edilen hastalardan alınan klinik izolatlara karşı lopinavirin in-vitro aktivitesinin olduğu saptanmıştır. Nelfinavire (n = 13) ve sakuinavire (n = 4), 4 katından daha fazla azalmış duyarlılık gösteren izolatlar, lopinavire karşı 4 kattan daha az duyarlılık göstermiştir. İndinavire (n = 16) ve ritonavire (n = 3), 4 katından daha fazla azalmış duyarlılık gösteren izolatlar, lopinavire karşı sırasıyla ortalama 5.7 ve 8.3 katlık bir azalmış duyarlılık göstermiştir. Daha evvel iki ya da daha fazla proteaz inhibitörü ile tedavi edilen hastalardan alınan izolatlar, klinik kısımda tarif edildiği üzere (bkz. Klinik çalışmalar: Önceden proteaz inhibitör tedavisi almış hastalarda KALETRA®’nın antiviral aktivitesi) lopinavire karşı duyarlılıkta daha fazla azalma görülmüştür.
Lopinavir/ritonavir tedavisi esnasında çapraz direnç
Lopinavir/ritonavir tedavisi esnasında seçilen virüslerde çapraz dirence ilişkin çok az bilgi bulunmaktadır.
Daha önce bir veya daha fazla proteaz inhibitörüyle tedavi edilmiş olan 4 hastanın Lopinavir/ritonavir tedavisi sırasında fenotipik lopinavir direnci gelişmiş olan izolatları ya çapraz-dirençli kalmış ya da ritonavir, indinavir ve nelfinavire çapraz direnç geliştirmişlerdir. Bütün rebound virüsler ya tümüyle duyarlı olarak kalmış ya da amprenavire karşı orta derecede azalmış duyarlılık gelişmiştir (lopinavire 99 kat direnç ile eşzamanlı olarak 8.5 kata kadar). Daha önce sakinavir tedavisi uygulanmamış iki bireyden elde edilen rebound izolatlar sakinavire karşı tümüyle duyarlı kalmıştır.
Lopinavir/ritonavire dayalı bir kombinasyon rejimi başlatılan antiretroviral deneyimi olan hastalarda azalmış virolojik yanıtın genotipik ilişkisi
Lopinavir/ritonavire virolojik yanıt, başlangıç noktasındaki proteazlardaki şu üç ya da daha fazla amino asit substitüsyonunun varlığı ile lopinavir/ritonavire virolojik yanıtın etkilendiği gösterilmiştir: L10F/I/R/V, K20M/N/R, L24I, L33F, M36I, 147V, G48V, I54L/T/V, V82A/F/S/T ve I84V. Tablo 1, Çalışma 888, 765 ve 957’de başlangıç noktasındaki yukarıdaki proteaz inhibitörüne direnç mutasyonlarının sayısına göre 48 haftalık virolojik yanıtı (HIV RNA < 400 kopya/ml) gösterir (aşağıya bakınız).
Tablo 1
Başlangıç noktasına göre Lopinavir/ritonavir Duyarlılığı ve Lopinavir/ritonavireazalmış yanıt ile ilişkilendirilen Proteaz Substitüsyonlarının sayısında göre 48. haftada Virolojik Yanıt (HIV RNA < 400 kopya /ml) 1
Başlangıç noktasındaki proteaz inhibitörü mutasyonlarının sayısı1 | Çalışma 888 (Tek proteaz inhibitörü deneyimli, NNRTI kullanmamış)2 n = 130 | Çalışma 765 (Tek proteaz inhibitörü deneyimli, NNRTI kullanmamış) 3 n = 56 | Çalışma 957 (çoklu proteaz inhibitörü deneyimli, NNRTI kullanmamış) 4 n = 50 |
0 - 2 | 76/103 (74%) | 34/45 (76%) | 19/20 (95%) |
3 - 5 | 13/26 (50%) | 8/11 (73%) | 18/26 (69%) |
6 ya da daha fazla | 0/1 (0%) | - | 1/4 (25%) |
1 Analizde yer alan substitüsyonlar: L10F/I/R/V, K20M/N/R, L24I, | L33F, M36I, I47V, | ||
G48V, I54L/T/V, V82A/C/F/S/T ve I84V.
2 43% indinavir, 42% nelfinavir, 10% ritonavir, 15% sakinavir.
3 41% indinavir, 38% nelfinavir, 4% ritonavir, 16% sakinavir.
4 86% indinavir, 54% nelfinavir, 80% ritonavir, 70% sakinavir.
Klinik çalışmalar
Önceden proteaz inhibitör tedavisi almış hastalarda KALETRA®’nın antiviral aktivitesi:
Lopinavire karşı in vitro duyarlılık azalmasının klinik anlamı, başlangıçtaki viral genotipe ve fenotipe göre Kaletra tedavisine virolojik yanıt değerlendirilerek, daha önce nelfinavir, indinavir, sakinavir ve ritonavirden (Çalışma M98-957) seçilen en az 2 proteaz inhibitörüyle tedaviye rağmen NNRTI kullanmamış ve HIV RNA > 1000 kopya/ml sahip 56 hastada incelenmiştir. Bu çalışmada hastalar başlangıç olarak efavirenz ve nükleozid ters transkriptaz inhibitörleri ile kombinasyon halinde lopinavir/ritonavirin iki dozundan birini almak için randomize edildi. Lopinavirin başlangıç noktasındaki 56 viral izolata karşı EC50 değeri, yabanıl tipteki HIV’e karşı EC50 değerine göre 0.5 ila 96 kat daha yüksektir. Bu başlangıç noktasındaki izolatların %56’sı (31/56), lopinavire karşı 4 kat daha fazla azalmış duyarlılık göstermiştir. 31 izolatın lopinavir duyarlılığında 27.9 katlık bir ortalama azalma bulunmakta idi.
KALETRA®, efavirenz ve nükleozid ters transkriptaz inhibitörleriyle ile 48 haftalık tedaviden sonra, lopinavire karşı < 10-kat, 10 ila 40-kat arası ve > 40-kat duyarlılık azalması olan hastaların sırasıyla %93 (25/27), %73 (11/15) ve %25’inde (2/8) plazma HIV RNA’nın < 400 kopya/ml olduğu gözlemlenmiştir. Lopinavir duyarlılığı rekombinant fenotipik teknolojisi kullanılarak virolojik olarak test edilmiştir. Genotip de ayrıca virolojik olarak yapılmıştır. Plazma HIV RNA < 50 kopya/ml, sırasıyla yukarıdaki hasta gruplarının %81 (22/27), %60 (9/15) ve %25 (2/8)’inde gözlemlenmiştir
Şu an lopinavir/ritonavir tedavisi gören hastalardaki izolatlarda, lopinavirle ilişkili mutasyon örnekleri aydınlatmak için yeterli veri bulunmamaktadır. Spesifik mutasyon örnekleri ve virolojik yanıt oranları arasındaki ilişkiyi değerlendirmek için daha fazla çalışma gerekmektedir.
Klinik farmakodinamik veriler Erişkinlerdeki Kullanımı:
Önceden antiretroviral tedavi almayan hastalar
Çalışma M98-863: lopinavir/ritonavir günde 2 kez + stavudin + lamivudin ile nelfinavir (günde 3 kez) + stavudin
M98-863 çalışması, daha önce antiretroviral almamış olan 653 hastada nelfinavir (günde üç defa 750 mg) artı stavudin + lamivudinin ile lopinavir/ritonavir (günde iki defa 400/100 mg) artı stavudin + lamivudin tedavisi ile karşılaştırıldığı devam etmekte olan randomize, çift kör, çok merkezli bir çalışmadır. Yaş ortalaması 38 olan (aralık 19 ila 84 arası) hastaların %57’si beyaz ırka mensup ve %80’i erkek idi. Ortalama başlangıç noktasındaki CD4 hücresi sayısı 259 hücre/mm3 (aralık: 2 - 949 hücre/mm3) ve ortalama başlangıç noktasındaki plazma HIV-1 RNA 4.9 log10 kopya/ml (aralık: 2.6 - 6.8 log10 kopya/ml) idi.
Çalışma M97-765: Günde 2 kez lopinavir/ritonavir + nevirapin + NRTI’
M97-765 çalışması, iki doz düzeyinde (400/100 mg ve 400/200 mg, ikisi de günde iki defa) lopinavir/ritonavir artı nevirapin (günde iki defa 200 mg) ve iki nükleosid ters transkriptaz inhibitörünü, daha evvelden tek proteaz inhibitörü deneyimi olan ve daha önce nükleosid olmayan ters transkriptaz inhibitörü kullanmamış olan 70 hastada değerlendiren randomize, kör, çok merkezli devam etmekte olan bir çalışmadır. Yaş ortalaması 40 olan (aralık 22 ila 66 arası) hastaların %73’i beyaz ırka mensup ve %90’sı erkek idi. Ortalama—başlangıç noktasındaki CD4 hücre sayısı 372 hücre/mm3 (aralık 72 - 807 hücre/mm3) ve ortalama başlangıç noktasındaki plazma HIV-1 RNA 4.0 log10 kopya/ml (aralık 2.9 -5.8 log10 kopya/ml) olmuştur.
Çalışma 765’te tedavinin 144 haftası boyunca, HIV RNA < 400 (< 50) kopya/ml olan hastaların oranı %54 (%50) [n=70] ve CD4 hücre sayısında buna karşılık gelen ortalama artış 212 hücre/mm3 olmuştur. 27 hasta (%39) çalışmayı bırakmış olup advers olaylar nedeniyle bırakan 9 hasta (%13) ve 2 ölüm (%3) buna dahildir.
Pediyatrik Kullanım
Çalışma M98-940
Çalışma M98-940, 80 mg/ml lopinavir ve 20 mg/ml ritonvir içeren lopinavir/ritonavir oral solüsyonunun farmakokinetik profilini, tolerabilitesini, güvenliliğini ve etkinliğini daha önce antiretroviral kullanmamış (%44) ve antiretroviral deneyimli (%56) 100 pediyatrik hastada değerlendiren açık etiketli, çok merkezli bir çalışmadır. Bütün hastalar daha önce non-nükleozid ters transkriptaz inhibitörü kullanmamış olan hastalardır. Hastalar m2 başına 230 mg lopinavir/57.5 mg ritonavir dozu ya da m2 başına 300 mg lopinavir/75 mg ritonavir dozu alacak şekilde randomize edilmişlerdir. Daha önce kullanmamış hastalara ayrıca nükleosid ters transkriptaz inhibitörleri verilmiştir. Deneyimli hastalar nevirapin artı en fazla iki nükleozid ters transkriptaz inhibitörü almışlardır.
5.2. Farmakokinetik özellikler
Genel özellikler
Ritonavir ile birlikte verilen lopinavirin farmakokinetik özellikleri sağlıklı yetişkin gönüllülerde ve HIV ile enfekte hastalarda değerlendirilmiştir: iki grup arasında önemli farklar gözlenmemiştir. Lopinavir esasen tümüyle CYP3A tarafından metabolize edilir. Ritonavir, lopinavir metabolizmasını inhibe ederek lopinavirin plazma düzeylerini arttırır. Çalışmalarda lopinavir/ritonavirin günde iki defa 400/100 mg dozunda kullanımı HIV ile enfekte hastalarda ritonavirden 15-20 kat daha yüksek olan ortalama kararlı durum lopinavir plazma konsantrasyonları elde edilmiştir. Ritonavirin plazma düzeyleri, günde iki defa 600 mg ritonavir dozundan sonra elde edilen düzeylerden %7 daha azdır. Lopinavirin in vitro antiviral EC50 değeri, ritonavirinkinden yaklaşık 10 kat daha düşüktür. Bu nedenle, KALETRA®’nın antiviral aktivitesi lopinavire bağlıdır.
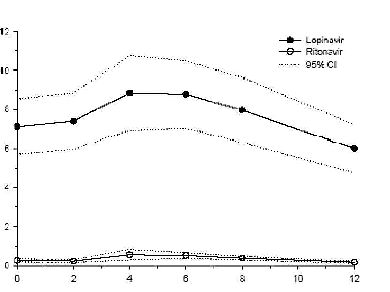
Şekil 1, HIV-enfekteli yetişkin hastalarda (n = 19) yapılan bir farmakokinetik çalışmadaki yemeklerle birlikte üç hafta boyunca günde iki defa 400/100 mg lopinavir/ritonavir uygulaması sonrası lopinavirin ve ritonavirin ortalama kararlı durum plazma konsantrasyonlarını gösterir.
Absorpsiyon: HIV-pozitif hastalardaki bir farmakokinetik çalışmada, 3 hafta süreyle yemeklerle birlikte günde iki defa 400/100 mg KALETRA® ile çoklu dozlama, uygulamadan ortalama 4 saat sonra gelişen 9.8 ± 3.7 mcg/ml’lik bir ortalama ± SD lopinavir doruk plazma konsantrasyonu (Cmax) meydana getirir. Sabah dozundan önceki ortalama kararlı durum vadi konsantrasyonu 7.1 ± 2.9 mcg/ml ve 12 saatlik bir doz aralığı boyunca lopinavir EAA’sı ortalama 92.6 ± 36.7 ^g^saat/ml olmuştur. Ritonavirle birlikte formüle edilen lopinavirin insanlardaki mutlak biyoyararlanımı belirlenmiş değildir.
Dağılım: Kararlı durumda, lopinavir plazma proteinlerine yaklaşık %98-99 oranında bağlanır. Lopinavir, alfa-1-asit glikoprotein (AAG) ve albümine bağlanır ama AAG için daha yüksek bir afiniteye sahiptir. Kararlı durumda lopinavirin proteine bağlanması, günde iki defa 400/100 mg KALETRA® dozundan sonra gözlemlenen konsantrasyon aralığı boyunca sabit kalmakta olup sağlıklı gönüllülerde ve HIV-pozitif hastalarda benzer düzeydedir.
Metabolizma: İnsan karaciğer mikrozomlarıyla yapılan in vitro deneyler, lopinavirin başlıca oksidatif metabolizmaya tabi olduğunu göstermiştir. Lopinavir yaygın olarak karaciğer sitokrom P450 sistemince neredeyse tümüyle CYP3A izoenzimi tarafından metabolize edilir. Ritonavir güçlü bir CYP3A inhibitörü olup lopinavir metabolizmasını inhibe ederek lopinavirin plazma düzeylerini artırmaktadır. İnsanlarda yapılan bir 14C-lopinavir çalışması, tek doz 400/100 mg KALETRA® dozundan sonra plazmadaki radyoaktivitenin %89’unun ana ilaca bağlı olduğunu göstermiştir. İnsanda lopinavirin en az 13 oksidatif metaboliti saptanmıştır. 4-okso ve 4-hidroksimetabolit epimerik çifti antiviral aktiviteye sahip majör metabolitlerdir ama bunlar toplam plazma radyoaktivitesinin yalnızca küçük miktarlarını oluşturmaktadır. Ritonavirin metabolik enzimleri indükleyerek kendi metabolizmasını ve benzer biçimde lopinavir metabolizmasını indüklediği gösterilmiştir. Çoğul doz sırasında doz öncesi lopinavir konsantrasyonları, zamanla düşerek yaklaşık 10 - 16 gün sonra stabilize olmaktadır.
Şekil 1
*HIV-enfekteli Yetişkin Deneklere ait %95 Güven aralıklarına (CV) sahip Ortalama kararlı durum plazma konsantrasyonları (N=19)
H
n
o
y
s astr
n o
a
Zaman (saat)
Eliminasyon: 400/100 mg 14C-lopinavir/ritonavir dozunu takiben, uygulanan 14C-lopinavir dozunun sırasıyla yaklaşık %10.4 ± %2.3 ve %82.6 ± %2.5’i, 8 gün sonra idrar ve dışkıda bulunmaktadır. Değişmemiş lopinavir idrar ve dışkıda, verilen dozun sırasıyla %2.2 ve %19.8’i oranında bulunmaktadır. Çoklu dozlardan sonra lopinavir dozunun %3’ten azı değişmeden idrarla atılmaktadır. Lopinavirin görünen oral klerensi (CL/F) 5.98 ± 5.75 L/saat (ortalama ± Standart sapma, N=19) olmuştur.
Günde bir kez doz uygulaması
Günde bir kez lopinavir/ritonavir uygulamasının farmakokinetiği özellikleri HIV-enfekte daha önce antiretroviral tedavi görmemiş hastalarda değerlendirilmiştir. Lopinavir/ritonavir 800/200 mg, günde bir kez doz rejiminin bir parçası olarak emtrisitabin 200 mg ve tenofovir DF 300 mg ile kombinasyon halinde uygulandı. 800/200 mg lopinavir/ritonavirin günde bir kez olarak 4 hafta boyunca yemeklerle birlikte çoklu doz uygulaması (n = 24), uygulamadan yaklaşık 6 saat sonra gelişen 11.8 ± 3.7 ^g/ml’lik bir ortalama ± standart sapma lopinavir doruk plazma konsantrasyonu (Cmaks) oluşturur. Lopinavirin ortalama kararlı durum vadi konsantrasyonu sabah dozundan önce lopinavirin ortalama kararlı durum vadi konsantrasyonu 3.2 ± 2.1 |ig/ml ve bir doz aralığı içerisinde minimum konsantrasyonu 1.7 ± 1.6 ^g/ml’dir. 24 saatlik bir doz aralığı boyunca lopinavir EAA’sı ortalama 154.1 ± 61.4 ^g*h/ml. olmuştur.
Elektrokardiyogram üzerine etkisi:
QTcF aralığı 39 sağlıklı yetişkinde randomize, plasebo ve aktif (moksifloksasin 400 mg QD) kontrollü çapraz çalışmada, Gün 3’de 12 saatin üzerinde 10 ölçüm ile değerlendirilmiştir. Plasebodan gelen QTcF’deki en yüksek ortalama farklılıklar (%95 üst güven sınırı) günde iki kez 400/100 mg ve supraterapötik olarak günde iki kez 800/200 mg LPV/r için sırasıyla 3.6 (6.3) ve 13.1 (15.8) idi.Bu iki doz rejimi Gün 3’de aşağı yukarı tavsiye edilen QD ile birlikte gözlenenden veya kararlı durumda günde iki defa LPV/r’den 1.5 ve 3 kat fazla açığa çıkma ile sonuçlandı. Hiç bir denek temel seviyeden > 60 msaniyenin QTcF’sinde artış veya QTcF aralığında 500 msaniye’lik potansiyel klinik olarak uygun eşik değerinin üstüne çıkma durumunu tecrübe etmemiştir.
PR aralığının hafif uzaması aynı çalışmada Gün 3’de lopinavir/ritonavir alan deneklerde tespit edilmiştir. En yüksek PR aralığı 286 msaniye olup herhangi ikincil veya üçüncül derecede kalp bloğu gözlenmemiştir. (bakınız bölüm 4.4)
Hastalardaki Karakteristik Özellikler
Pediyatrik hastalar: Günde iki defa 300/75 mg/m2 ve 230/57.5 mg/m2 lopinavir/ritonavir dozlarının farmakokinetiği, yaşları altı ay ile 12 yaş arasında değişen toplam 53 pediyatrik hastada yapılan çalışmalarda incelenmiştir. Nevirapinsiz günde iki defa 230/57.5 mg/m2 rejimi ve nevirapinle günde iki defa 300/75 mg/m2 rejimi, günde iki defa 400/100 mg, (nevirapinsiz) yetişkin hasta rejimindekine benzer lopinavir plazma konsantrasyonları sağlamıştır. Günde bir kez lopinavir/ritonavir uygulaması pediyatrik hastalarda değerlendirilmemiştir.
Günde iki defa 230/57.5 mg/m2 lopinavirle ortalama kararlı durum EAA, Cmax ve Cmin değerleri nevirapin olmadan lopinavir/ritonavirden sonra (n=12) sırasıyla 72.6 ± 31.1mcg*saat/ml, 8.2 ± 2.9 ve 3.4 ± 2.1 mcg/ml ve günde iki defa 300/75 mg/m2 ile (nevirapinle beraber) (n=12) 85.8 ± 36.9 mcg*saat/ml, 10.0 ± 3.3 ve 3.6 ± 3.5 mcg/ml olmuştur. Nevirapin rejimi, günde iki kez 7 mg/kg (6 ay ila 8 yaş arası için) ya da günde iki kez 4 mg/kg’dır (8 yaşın üzeri için).
Altı aydan küçük pediyatrik hastalarda KALETRA®’nın güvenliliği ve farmakokinetik profilleri kanıtlanmamıştır. Bir klinik çalışma esnasında altı ay ile 12 yaş arası HIV-enfekteli hastalarda görülen advers olay profili yetişkin hastalarınkine benzer idi. Klinik çalışmalarda KALETRA®’nın pediyatrik hastalardaki antiviral aktivitesinin değerlendirmesi devam etmektedir. Günde bir kez KALETRA® uygulaması pediyatrik hastalarda değerlendirilmemiştir.
Böbrek yetmezliği: Lopinavirin farmakokinetiği, böbrek yetmezliği olan hastalarda araştırılmamıştır; ancak, lopinavirin böbrek klerensi ihmal edilebilir olduğundan, böbrek yetmezliği olan hastalarda toplam vücut klerensinde bir azalma olması beklenmemektedir.
Karaciğer yetmezliği: Lopinavir çoğunlukla karaciğerde metabolize edilir ve atılır. Hafif veya orta dereceli karaciğer yetmezliği olan HIV ve HCV ile kombine enfeksiyonlu hastalarda, günde iki defa lopinavir/ritonavir 400/100 mg ile yapılan çoğul dozlu bir çalışmasında, normal karaciğer fonksiyonuna sahip HIV enfeksiyonlu hastalara kıyasla lopinavirin EAA’sında %30’luk bir artış ve Cmaks’ında %20’lik azalma olmuştur. Buna ek olarak, lopinavirin plazma proteinlerine bağlanması, kontrol grubuna kıyasla hafif ila orta düzeyde karaciğer yetmezliği olanlarda daha azdır (sırasıyla 99.09 ’e karşı. 99.31%) (bkz. 4.5.).
Cinsiyet, Yaş, Irk,
5.3. Klinik öncesi güvenlilik verileri
Akut, Subakut ve Kronik Toksisite
Kemirgenlerde ve köpeklerdeki yinelenen dozlu toksisite çalışmaları, majör hedef organların karaciğer, böbrek, tiroid, dalak ve dolaşımdaki eritrositler olduğunu göstermiştir. Hepatik değişimler fokal dejenerasyonla birlikte hücre şişmesidir. Bu değişimlere yol açan maruziyet, insandaki klinik maruziyet ile karşılaştırılırken hayvanlardaki dozajlar önerilen klinik dozun 6 katından fazladır. İnsanda önerilen dozun en az iki katını alan farelerde hafif renal tübüler dejenerasyon görülürken sıçanlarda ve köpeklerde böbrekler etkilenmemiştir. Sıçanlarda serum tiroksinindeki azalma, tiroid bezlerinde foliküler hücre hipertrofisi ile sonuçlanan bir TSH salınımı artışına yol açmıştır. Bu değişimler etkin maddenin kesilmesiyle geri dönüşlü olup farelerde ve köpeklerde görülmemiştir. Sıçanlarda Coombs-negatif anizositoz ve poikilositoz gözlenmiş ama farelerde ve köpeklerde gözlenmemiştir. Histiyositoz ile birlikte dalak büyümesi sıçanlarda görülmüş ama diğer türlerde görülmemiştir. Serum kolesterolü kemirgenlerde yükselmiş ama köpeklerde yükselmemi ştir. Trigliseridler yalnızca farelerde yükselmiştir.
6. FARMASÖTİK ÖZELLİKLER
6.1. Yardımcı maddelerin listesi
Tablet içeriği:
Kopovidon Sorbitan laurat Kolloidal anhidr silika Sodyum stearil fumarat
Film kaplama
Hipromelloz Titanyum dioksit
Makrogol tip 400 (Polietilen glikol 400) Hidroksipropil selüloz Talk
6.2. Geçimsizlikler
Uygulanamaz.
6.3. Raf ömrü
6.4. Saklamaya yönelik özel tedbirler
6.5. Ambalajın niteliği ve içeriği
6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemler
Kullanılmamış olan ürünler yada atık materyaller ’Tıbbi ürünlerin kontrolü yönetmeliği’ve ’Ambalaj atıklarının kontrolü yönetmelikleri’ne uygun olarak imha edilmelidir.
 Sırt Ağrısı
Sırt ağrısı birden bire ortaya
çıkıp şiddetli (akut) olabilir veya zamanla gelişip daha uzun
süreli sorunlara (kronik) neden olabilir.
Sırt Ağrısı
Sırt ağrısı birden bire ortaya
çıkıp şiddetli (akut) olabilir veya zamanla gelişip daha uzun
süreli sorunlara (kronik) neden olabilir. |
 Asperger Sendromu
Asperger sendromu, otistik gurubun bir bölümü olan bir özürdür. Bu genelde,
gurubun daha ”yüksek” tarafında yer aldığı düşünülen kişilere uygun bir tanıdır.
Asperger Sendromu
Asperger sendromu, otistik gurubun bir bölümü olan bir özürdür. Bu genelde,
gurubun daha ”yüksek” tarafında yer aldığı düşünülen kişilere uygun bir tanıdır. |
İLAÇ EŞDEĞERLERİ
| Eşdeğer İlaç Adı | Barkodu | İlaç Fiyatı |
|---|---|---|
| ORVICAL | 8680199093704 | 6,935.56TL |
| Diğer Eşdeğer İlaçlar |
 |
Artrit Artrit, oldukça yaygın bir hastalıktır ancak iyi anlaşılamamıştır. Aslında “artrit” tek bir hastalığın adı değildir; eklem ağrısı veya eklem hastalıklarını adlandırmanın gayri resmi yoludur. |
 |
Ağız Kanseri Ağız kanserinin en yaygın türleri, dudak, dil, dişetidir. Nadiren yanak içi veya damak bölgelerini de içine alır. |
 |
HIV ve Aids HIV, Human Immunodeficiency Virus’dür (İnsanlarda Bağışıklık Sistemini Bozan Virüsdür). Bu virüs AIDS hastalığına sebep olur. |
İLAÇ GENEL BİLGİLERİ
Abbott Laboratuvarları İthalat İhracat Tic. Ltd. Şti.
| Geri Ödeme Kodu | A10816 |
| Satış Fiyatı | TL |
| Önceki Satış Fiyatı | |
| Original / Jenerik | Original İlaç |
| Reçete Durumu | Normal Reçeteli bir ilaçdır. |
| Barkodu | 8699548093692 |
| Etkin Madde | Lopinavir + Ritonavir |
| ATC Kodu | J05AR10 |
| Birim Miktar | 200+50 |
| Birim Cinsi | MG |
| Ambalaj Miktarı | 120 |
| Enfeksiyona Karşı Kullanılan (Antienfektif) İlaçlar > Virüslere Karşı Direkt Etkili İlaçlar > Lopinavir ve Ritonavir |
| İthal ( ref. ülke : Ingiltere ) ve Beşeri bir ilaçdır. |