HEMLIBRA 30 mg/ 1 ml SC enjeksiyonluk ��zelti K�sa �r�n Bilgisi
{ Emisizumab }
1. BE�ER� TIBB� �R�N�N ADI
HEMLIBRA 30 mg/1 mL S.C. enjeksiyonluk ��zelti Steril
2. KAL�TAT�F VE KANT�TAT�F B�LE��M
Etkin madde
1 mL'lik her bir flakon, 30 mg/mL konsantrasyonda 30 mg emicizumab i�ermektedir.
Emicizumab; �in Hamsteri Over (CHO) h�crelerinde rekombinant DNA teknolojisi ile �retilmi�, fakt�r IXa ve fakt�r X'u birle�tiren, bispesifik antikor yap�s�na sahip bir monoklonal, h�manize, modifiye edilmi� imm�noglobulin G4 (IgG4) antikorudur.
Yard�mc� maddeler
Yard�mc� maddeler i�in B�l�m 6.1'e bak�n�z.
3. FARMAS�T�K FORMU
Enjeksiyonluk ��zelti i�eren flakon. Renksiz ile hafif sar� aras� renkte ��zelti.
4. KL�N�K �ZELL�KLER
4.1. Terap�tik endikasyonlar
HEMLIBRA, fakt�r VIII inhibit�rl� ya da inhibit�rs�z hemofili A (konjenital fakt�r VIII eksikli�i) hastalar�nda rutin profilakside endikedir.
HEMLIBRA t�m ya� gruplar�nda kullan�labilir.
4.2. Pozoloji ve uygulama �ekli
Ba�ka bir biyolojik t�bbi �r�nle de�i�tirilmesi, re�eteyi yazan hekimin onay�n� gerektirmektedir.
Tedavi, hemofili ve/veya kanama bozukluklar�n�n tedavisinde deneyimli bir hekimin g�zetiminde ba�lat�lmal�d�r.
Bypass ajanlar�yla (�rn. aPCC ve rFVIIa) tedavi, (rutin profilaksi dahil) HEMLIBRA tedavisi ba�lat�lmadan �nceki g�n durdurulmal�d�r (bkz. B�l�m 4.4).
Fakt�r VIII (FVIII) profilaksisine HEMLIBRA tedavisinin ilk 7 g�n�nde devam edilebilir.
Pozoloji/uygulama s�kl��� ve s�resi:
�nerilen doz, subkutan enjeksiyon yolu ile ilk 4 hafta boyunca haftada 1 kez 3 mg/kg (y�kleme dozu) ve bunu takiben idame dozu olarak haftada 1 kez 1,5 mg/kg, 2 haftada 1 kez 3 mg/kg veya 4 haftada 1 kez 6 mg/kg'd�r ve t�m dozlar subkutan enjeksiyon olarak verilir.
Y�kleme dozu, idame dozundan ba��ms�z olarak ayn�d�r.
Uyuncu desteklemek ad�na, idame doz rejimi hekim ve hasta/hasta bak�m�n� �stlenen ki�inin tercihine g�re se�ilmelidir.
Uygulama �ekli:
Hasta dozu (mg cinsinden) ve hacmi (mL cinsinden) a�a��daki �ekilde hesaplanmal�d�r:
�lk 4 hafta boyunca haftada 1 kez y�kleme dozu (3 mg/kg):
Hasta v�cut a��rl��� (kg) x doz (3 mg/kg) = uygulanacak toplam emicizumab miktar� (mg)
Sonras�nda idame dozu olarak 5. haftadan itibaren haftada 1 kez 1,5 mg/kg, 2 haftada 1 kez 3 mg/kg veya 4 haftada bir kez 6 mg/kg:
Hasta v�cut a��rl��� (kg) x doz (1,5; 3 veya 6 mg/kg) = uygulanacak toplam emicizumab miktar� (mg)
Subkutan olarak enjekte edilecek toplam HEMLIBRA hacmi a�a��daki �ekilde hesaplan�r:
Uygulanacak toplam emicizumab miktar� (mg) ÷ flakon konsantrasyonu (mg/mL) = enjekte edilecek toplam HEMLIBRA hacmi (mL).
Uygulanacak toplam hacim haz�rlan�rken farkl� HEMLIBRA konsantrasyonlar� (30 mg/mL ve 150 mg/mL) birle�tirilmemelidir.
Enjeksiyon ba��na 2 mL'nin �zerinde hacim uygulanmamal�d�r. �rnekler:
Hastan�n v�cut a��rl��� 16 kg, hastaya uygulanan idame doz rejimi haftada 1 kez 1,5 mg/kg:
Y�kleme dozu (ilk 4 hafta) �rne�i: 16 kg x 3 mg/kg = 48 mg y�kleme dozu i�in gerekli emicizumab miktar�.
Uygulanacak hacmi hesaplamak �zere hesaplanm�� doz 48 mg, 150 mg/mL'ye b�l�n�r; 48 mg emicizumab ÷ 150 mg/mL = 0,32 mL enjekte edilecek 150 mg/mL HEMLIBRA konsantrasyonu.
4.3. Kontrendikasyonlar
Etkin madde
4.4. �zel kullan�m uyar�lar� ve �nlemleri
�zlenebilirlik
Biyolojik t�bbi �r�nlerin izlenebilirli�ini artt�rmak �zere, uygulanan �r�n�n ad� ve seri numaras� net olarak kaydedilmelidir.
HEMLIBRA ve aktive protrombin kompleksi konsantresi (aPCC) ile ba�lant�l� trombotik mikroanjiyopati
HEMLIBRA profilaksisi alan hastalarda y�r�t�len bir klinik �al��mada, 24 saat veya daha uzun s�reyle >100U/kg/24 saat ortalama k�m�latif miktarda aktive protrombin kompleks konsantresi (aPCC) uyguland��� durumda trombotik mikroanjiopati (TMA) vakalar� bildirilmi�tir (bkz. B�l�m 4.8). TMA olaylar�na y�nelik tedavi, plazmaferez veya hemodiyaliz ile birlikte ya da tek ba��na destekleyici tedavi uygulanmas�n� kapsamaktad�r. HEMLIBRA tedavisine ara verilmesi ve aPCC'nin kesilmesinin ard�ndan bir hafta i�erisinde TMA'n�n iyile�ti�i g�r�lm��t�r. Bu h�zl� klinik iyile�me, trombotik trombositopenik purpura ve atipik hemolitik �remik sendrom gibi klasik TMA'larda g�zlemlenen ola�an klinik seyirden farkl�d�r (bkz. B�l�m 4.8). Bir hasta TMA'n�n d�zelmesini takiben HEMLIBRA'ya yeniden ba�lam�� ve tedavisi g�venli bir �ekilde s�rd�r�lm��t�r.
HEMLIBRA profilaksisi alan hastalara aPCC uygulan�rken TMA geli�imi a��s�ndan hastalar�n izlenmesi gereklidir. Hekim, TMA ile tutarl� klinik semptomlar�n ve/veya laboratuvar bulgular�n�n g�zlenmesi durumunda aPCC'yi acilen b�rakmal�, HEMLIBRA tedavisine ara vermeli ve klinik olarak endike oldu�u �ekilde tedavi uygulamal�d�r. Hekimler ve hastalar/bak�m verenler, her bir vaka i�in ayr� ayr� olmak �zere, TMA'n�n ortadan kalkmas�n� takiben HEMLIBRA profilaksisini yeniden ba�latman�n faydalar�n� ve risklerini de�erlendirmelidir. HEMLIBRA profilaksisi uygulanan bir hastada bir bypass edici ajan�n endike oldu�u durumda, a�a��da bulunan bypass edici ajanlar�n kullan�m�na y�nelik doz uygulamas� k�lavuzuna bak�n�z.
Y�ksek TMA riski (�rn. TMA ge�mi�i veya ailesel TMA ge�mi�i) olan veya TMA geli�me risk fakt�r� bulunan kombine ila� (�rn. siklosporin, kinin, takrolimus) kullanan hastalar�n tedavisinde dikkatli olunmal�d�r.
HEMLIBRA ve aktive protrombin kompleksi konsantresi (aPCC) ile ba�lant�l� tromboembolizm
HEMLIBRA profilaksisi alan hastalarda y�r�t�len bir klinik �al��mada, 24 saat veya daha uzun s�re >100U/kg/24 saat ortalama k�m�latif miktarda aktive protrombin kompleksi konsantresi (aPCC) uyguland���nda trombotik olaylar�n (TE) ortaya ��kt��� bildirilmi�tir (bkz. B�l�m 4.8). Hi�bir vaka antikoag�lan tedavisini gerektirmemi�tir. aPCC'nin b�rak�lmas� ve HEMLIBRA tedavisinin kesilmesini takiben, iyile�me veya d�zelme kan�t� bir ay i�inde g�r�lm��t�r (bkz. B�l�m 4.8). Bir hasta trombotik olay�n d�zelmesini takiben HEMLIBRA'ya yeniden ba�lam�� ve tedavi g�venli bir �ekilde s�rd�r�lm��t�r.
HEMLIBRA profilaksisi al�rken aPCC uygulanan hastalar tromboemboli geli�imi a��s�ndan izlenmelidirler. Hekim, trombotik olaylarla tutarl� klinik semptomlar, g�r�nt�leme ve/veya laboratuvar bulgular� ortaya ��karsa, aPCC'yi acilen b�rakmal�, HEMLIBRA tedavisine ara vermeli ve klinik olarak endike oldu�u �ekilde tedavi uygulamal�d�r. Hekimler ve hastalar/bak�m verenler, her bir vaka i�in ayr� ayr� olmak �zere, trombotik olaylar�n ortadan kalkmas�n� takiben HEMLIBRA profilaksisini yeniden ba�latman�n faydalar�n� ve risklerini de�erlendirmelidir. HEMLIBRA profilaksisi uygulanan bir hastada bir bypass edici ajan�n�n endike oldu�u durumda, a�a��da bulunan bypass edici ajanlar�n�n kullan�m�na y�nelik doz uygulamas� k�lavuzuna bak�n�z.
HEMLIBRA profilaksisi alan hastalarda bypass edici ajanlar�n kullan�m� ile ilgili k�lavuz Bypass edici ajanlarla tedavi HEMLIBRA tedavisinin ba�lat�ld��� g�nden �nce kesilmelidir.
Hekimler, HEMLIBRA profilaksisi uygulan�rken, gerekmesi durumunda, t�m hastalar� ve/ veya bak�m verenleriyle, bypass edici ajanlar�n kesin dozlar� ve uygulama s�kl�klar�n� tart��mal�d�r.
aPCC'nin 50 U/kg'a kadar olan ba�lang�� dozu ile kontrol edilemezse, t�bbi k�lavuzluk veya g�zetim alt�nda, TMA veya tromboembolizm te�hisi i�in laboratuvar izlemesi ve dozun tekrarlanmas�ndan �nce kanama kontrol� de g�z �n�nde bulundurularak, ek aPCC dozlar� uygulanmal�d�r. Toplam aPCC dozu, tedavinin ilk 24 saatinde 100 U/kg'� ge�memelidir. Tedaviyi uygulayan hekimler, ilk 24 saat i�inde 100 U/kg'u a�an aPCC tedavisini d���n�yorlarsa, TMA ve TE riskinin kanama riskine kar�� dikkatli bir �ekilde tart�lmas� gereklidir.
Klinik �al��malarda, HEMLIBRA profilaksisi uygulanan hastalarda tek ba��na aktive rekombinant FVII (rFVIIa) kullan�m� ile TMA ya da TE vakalar� g�zlenmemi�tir.
Bypass edici ajanlara ili�kin doz uygulama k�lavuzu, HEMLIBRA profilaksisinin b�rak�lmas�n� takiben en az 6 ay boyunca takip edilmelidir (bkz. B�l�m 5.2).
�mm�nojenisite
Klinik �al��malarda, emicizumab konsantrasyonunun azalmas�na yol a�an ve etkinlik kayb�na yol a�an n�tralize edici anti-emicizumab antikorlar�n�n geli�imi nadiren g�zlenmi�tir (bkz. B�l�m 4.8 ve 5.1). Klinik etkililik kayb� belirtileri olan hastalar (�rn. ani kanama olaylar�nda art��), etiyolojiyi belirlemek i�in derhal de�erlendirilmeli ve n�tralize edici anti-emicizumab antikorlar�ndan ��pheleniliyorsa di�er terap�tik se�enekler d���n�lmelidir.
HEMLIBRA'n�n koag�lasyon testlerine etkisi
HEMLIBRA eksik olan aktive fakt�r VIII'in (FVIIIa) tenaz kofakt�r� aktivitesini yerine koyar. �ntrinsik p�ht�la�ma temelli koag�lasyon laboratuvar testleri, aktive edilmi� p�ht�la�ma zaman� (ACT), aktive edilmi� parsiyel tromboplastin zaman� (�rn., aPTT) dahil, trombin yoluyla FVIII'in FVIIIa'ya aktive edilmesi i�in gerekli zaman dahil olmak �zere toplam p�ht�la�ma s�resini �l�er. Bu tip intrinsik yola�a dayal� testler, trombin ile aktivasyonu gerekmeyen HEMLIBRA ile a��r� derecede k�salm�� p�ht�la�ma s�releri verecektir. A��r� k�salm�� intrinsik p�ht�la�ma s�resi, aPTT'ye dayanan, tek a�amal� FVIII aktivite tayini gibi tek fakt�rl� tayinlerin t�m�n� bozacakt�r (bkz. B�l�m 4.4, Tablo 1). Bununla birlikte, kromojenik veya imm�no-bazl� y�ntemleri kullanan tek fakt�rl� analizler HEMLIBRA'dan etkilenmez ve a�a��da tarif edildi�i gibi FVIII kromojenik aktivite tayinleri i�in �zel hususlar ile birlikte tedavi esnas�nda koag�lasyon parametrelerini izlemek i�in kullan�labilir.
Kromojenik fakt�r VIII aktivite testleri, insan veya bovin koag�lasyon proteinleri ile birlikte �retilebilir. �nsan koag�lasyon fakt�rlerini i�eren tayinler HEMLIBRA'ya duyarl�d�r; fakat HEMLIBRA'n�n klinik hemostatik potansiyelini oldu�undan daha y�ksek g�sterebilir. Buna kar��l�k, bovin koag�lasyon fakt�rleri i�eren tayinler HEMLIBRA'ya duyarl� de�ildir (aktivite �l��lmez) ve endojen ya da inf�ze fakt�r VIII aktivitesinin izlenmesi ya da anti- FVIII inhibit�rlerinin �l��lmesi i�in kullan�labilirler.
HEMLIBRA, fakt�r VIII'e kar�� geli�en inhibit�rlerin varl���nda aktif kalmaktad�r ve bu nedenle fakt�r VIII inhibit�rleri i�in p�ht�la�ma bazl� Bethesda analizlerinde yanl��-negatif bir sonu� olu�turacakt�r. Bunun yerine, HEMLIBRA'ya duyars�z olan bovin temelli bir kromojenik fakt�r VIII tayini uygulanan bir kromojenik Bethesda tayini kullan�labilir.
Bu iki farmakodinamik belirte� in vivo HEMLIBRA'n�n ger�ek hemostatik etkisini yans�tmamakla birlikte (aPTT a��r� k�sal�r ve bildirilen fakt�r VIII aktivitesi oldu�undan fazla hesaplanabilir), HEMLIBRA'n�n pro-koag�lan etkisine ili�kin g�receli bir g�sterge sa�lar.
�zetle, HEMLIBRA ile tedavi edilen hastalarda intrinsik yolak p�ht�la�ma bazl� laboratuar testi bulgular� aktivitesinin izlenmesi, fakt�r replasman� veya anti-koag�lasyon i�in doz uygulama tayini veya fakt�r VIII inhibit�rleri titrelerinin �l��lmesinde kullan�lmamal�d�r. �ntrinsik p�ht�la�ma bazl� laboratuar testleri kullan�l�rsa, bulgular�n�n yanl�� yorumlanmas� kanama epizotlar� ya�ayan hastalar�n yetersiz tedavisine yol a�abilece�inden, bu da potansiyel olarak �iddetli veya hayati risk ta��yan kanamalarla sonu�lanabilece�inden dikkat edilmelidir.
HEMLIBRA'dan etkilenen ve etkilenmeyen laboratuvar testleri a�a��daki Tablo 1'de g�sterilmektedir. Uzun yar�lanma �mr�nden dolay�, koag�lasyon analizleri �zerindeki bu etkiler son dozu takiben 6 aya kadar s�rebilir (bkz. B�l�m 5.2).
Tablo 1. HEMLIBRA'dan Etkilenen ve Etkilenmeyen Koag�lasyon Test Sonu�lar�
HEMLIBRA'dan Etkilenen Sonu�lar | HEMLIBRA'dan Etkilenmeyen Sonu�lar |
Aktive parsiyel tromboplastin s�resi (aPTT)
4.5. Di�er t�bbi �r�nler ile etkile�imler ve di�er etkile�im �ekilleri
HEMLIBRA ile yeterli ya da iyi kontroll� ila�-ila� etkile�im �al��malar� y�r�t�lmemi�tir.
Klinik deneyim, HEMLIBRA ve aPCC aras�nda bir ila� etkile�imi oldu�una i�aret etmektedir (bkz. B�l�m 4.4 ve 4.8).
Klinik �ncesi deneylere dayal� olarak, HEMLIBRA ile rFVIIa veya FVIII i�in bir hiperkoag�labilite olas�l��� mevcuttur. HEMLIBRA koag�lasyon potansiyelini artt�rmaktad�r, bu nedenle hemostaza eri�mek i�in gerekli rFVIIa veya FVIII dozu, HEMLIBRA profilaksisi kullan�lmad��� zamana g�redahad���kolabilir.
Trombotik komplikasyon durumlar�nda hekim klinik endikasyonuna g�re rFVIIa veya FVIII kullan�m�n� kesmeyi ve HEMLIBRA profilaksisine ara vermeyi de�erlendirmelidir. Tedavinin daha ileri y�netimi i�in bireysel klinik duruma g�re ��z�m �retilmelidir.
Doz ayarlanmas� yap�l�rken, di�er ila�lar�n yar� �mr� d���n�lmelidir; �zellikle HEMLIBRA kullan�m�n�n kesilmesi durumunda hemen etki g�r�lmeyebilir.
FVIII kromojenik testlerinin kullan�lmas� koag�lasyon fakt�rlerinin uygulanmas�n� y�nlendirebilir ve trombofilik �zelliklerin test edilmesi de d���n�lebilir.
HEMLIBRA profilaksisi alan hastalarda anti fibrinolitiklerin aPCC veya rFVIIa ile birlikte kullan�m�na dair tecr�be s�n�rl�d�r. Ancak HEMLIBRA alan hastalarda, sistemik anti fibrinolitiklerin aPCC veya rFVIIa ile birlikte kullan�ld���nda trombotik olaylar�n geli�me ihtimali g�z �n�nde bulundurulmal�d�r.
�zel pop�lasyonlara ili�kin ek bilgiler Herhangi bir etkile�im �al��mas� yap�lmam��t�r. Pediyatrik pop�lasyon:
Listelenmi� etkile�imler hem yeti�kinler hem de �ocuklar i�in ge�erlidir.
4.6. Gebelik ve laktasyon
Gebelik kategorisi: C
�ocuk do�urma potansiyeli bulunan kad�nlar/Do�um kontrol� (Kontrasepsiyon)
HEMLIBRA kullanmakta olan ve �ocuk do�urma potansiyeline sahip kad�nlar, HEMLIBRA tedavisi s�resince ve tedavinin kesilmesini takiben en az 6 ay boyunca etkili do�um kontrol y�ntemleri kullanmal�d�rlar (bkz. B�l�m 5.2).
Gebelik d�nemi
Hayvanlar �zerinde yap�lan �al��malar, gebelik /ve-veya/ embriyonal/fetal geli�im /ve-veya/ do�um /ve-veya/ do�um sonras� geli�im �zerindeki etkiler bak�m�ndan yetersizdir (bkz. B�l�m 5.3). �nsanlara y�nelik potansiyel risk bilinmemektedir.
HEMLIBRA gerekli olmad�k�a gebelik d�neminde kullan�lmamal�d�r.
Gebe kad�nlarda HEMLIBRA kullan�m� ile ilgili herhangi bir klinik �al��ma bulunmamaktad�r. HEMLIBRA ile hayvanlarda �reme �al��malar� yap�lmam��t�r. HEMLIBRA'n�n gebe bir kad�na uyguland���nda fet�se zarar verip vermeyece�i veya �reme kapasitesini etkileyip etkilemeyece�i bilinmemektedir. HEMLIBRA, hamilelik s�ras�nda ve do�umdan sonra tromboz riskinin artt��� ve �e�itli gebelik komplikasyonlar�n�n artan Yayg�n Damar i�i P�ht�la�ma (YDP) riski le ili�kili oldu�u g�z �n�ne al�narak, yaln�zca anneye
y�nelik potansiyel yarar�n fetusa y�nelik potansiyel riskten daha fazla olmas� durumunda kullan�lmal�d�r.
Laktasyon d�nemi
Emicizumab�n anne s�t�ne ge�ip ge�medi�i bilinmemektedir. Emicizumab�n s�t �retimi �zerindeki etkisi ya da anne s�t�ndeki varl��� ile ilgili �al��ma y�r�t�lmemi�tir. �nsan IgG'nin insan s�t�nde bulundu�u bilinmektedir. Bebek i�in emzirmenin faydas� ve kad�n i�in tedavinin faydas� g�z �n�ne al�narak emzirmeyi b�rakma veya HEMLIBRA'y� b�rakma/uzak durma karar� verilmelidir.
�reme yetene�i/Fertilite
Hayvan �al��malar�, �reme toksisitesi a��s�ndan do�rudan veya dolayl� zararl� etkilere i�aret etmemektedir (bkz. B�l�m 5.3). �nsanlarda fertilite verileri mevcut de�ildir. Dolay�s�yla emicizumab�n erkek ve di�i fertilitesi �zerindeki etkisi bilinmemektedir.
4.7. Ara� ve makine kullan�m� �zerindeki etkiler
HEMLIBRA'n�n ara� ve makine kullan�m� �zerinde etkisi yoktur.
4.8. �stenmeyen etkiler
G�venlilik profilinin �zeti
HEML�BRA'n�n genel g�venlilik profili, klinik ara�t�rmalardan ve pazarlama sonras� g�zetimden elde edilen verilere dayanmaktad�r. HEMLIBRA ile ger�ekle�tirilen klinik �al��malardan bildirilen en ciddi advers ila� reaksiyonlar� (A�R'ler), kavern�z sin�s trombozu (CST) ve deri nekrozu ile e�zamanl� olarak ger�ekle�en y�zeyel ven trombozu dahil olmak �zere trombotik olaylar ve trombotik mikroanjiyopatidir (TMA) (bkz. a�a��daki k�s�m ve B�l�m 4.4).
En az bir doz HEMLIBRA ile tedavi edilmi� hastalar�n ≥%10'unda bildirilen en yayg�n A�R'ler: enjeksiyon yeri reaksiyonlar� (%20), artralji (%15) ve ba� a�r�s�d�r (%14).
HEMLIBRA profilaksisi uygulanan klinik �al��malarda toplamda �� hasta (%0,8) A�R'ler nedeniyle tedaviden �ekilmi�tir; bunlar TMA, y�zeyel tromboflebit ile e� zamanl� deri nekrozu ve ba� a�r�s�d�r.
Advers ila� reaksiyonlar�n�n tablo haline getirilmi� listesi
A�a��daki advers ila� reaksiyonlar� (A�R'ler), toplam 373 erkek hemofili A hastas�n�n rutin profilaksi olarak en az bir HEMLIBRA dozu ald��� pazarlama sonras� g�zetimden elde edilen verilere ve d�rt faz III klinik �al��man�n (yeti�kin ve ergen �al��malar� [BH29884 – HAVEN 1, BH30071 – HAVEN 3 ve BO39182 – HAVEN 4] ve pediyatrik �al��ma BH29992 – HAVEN 2) birle�tirilmi� verilerini temel al�r. Klinik ara�t�rma kat�l�mc�lar�ndan 266 hasta (%71) yeti�kindi, 47 hasta (%13) ergen (>12 ila <18 ya�), 55 hasta (%15) �ocuk (>2 ila < 12 ya�) ve 5 hasta (%1) bebektir (1 ay ila <2 ya�). �al��malar aras�nda medyan maruziyet s�resi 33 haftad�r (aral�k: 0,1 ila 94,3 hafta).
HEMLIBRA alm�� hastalar �zerinde ger�ekle�tirilen Faz III klinik �al��malardan ve pazarlama sonras� g�zetimdene ldee dilenA�R'ler,MedDRA sistem organ s�n�f�na g�re
listelenmektedir (Tablo 2). Her bir A�R i�in ilgili s�kl�k kategorileri a�a��daki s�n�fland�rmay� temel almaktad�r: �ok yayg�n (≤ 1/10), yayg�n (≥1/100 ila <1/10), yayg�n olmayan (≥1/1000 ila <1/100), seyrek (≥1/10.000 ila <1/1000), �ok seyrek (<1/10.000) ve bilinmiyor (mevcut verilere dayal� olarak tahmin edilemez).
Tablo 2 HEMLIBRA ile Yap�lan Birle�tirilmi� HAVEN Klinik �al��malar�nda ve Pazarlama Sonras� Deneyimde G�zlenen Advers �la� Reaksiyonlar�n�n �zeti
Sistem Organ S�n�f� | Advers reaksiyonlar (tercih edilen terim, MedDRA) | S�kl�k |
Kan ve lenf sistemi hastal�klar� | Trombotik mikroanjiyopati | Yayg�n olmayan |
Sinir sistemi hastal�klar� | Ba� a�r�s� | �ok yayg�n |
Vask�ler hastal�klar | Y�zeyel tromboflebit | Yayg�n olmayan |
Kavern�z sin�s trombozu | Yayg�n olmayan | |
Gastrointestinal hastal�klar | �shal | Yayg�n |
Deri ve deri alt� doku hastal�klar� | Deri nekrozu | Yayg�n olmayan |
Anjiyo�dem | Yayg�n olmayan | |
�rtiker | Yayg�n | |
D�k�nt� | Yayg�n | |
�skelet-kas ve ba� doku hastal�klar� | Artralji | �ok yayg�n |
Miyalji | Yayg�n | |
Genel rahats�zl�klar ve uygulama b�lgesine ili�kin hastal�klar | Enjeksiyon yeri reaksiyonu | �ok yayg�n |
Ate� | Yayg�n | |
Terap�tik yan�t azalmas� | Yayg�n olmayan |
Se�ilmi� advers ila� reaksiyonlar�n�n tan�m� Trombotik mikroanjiyopati
Havuzlanm�� faz III klinik �al��malar�nda, trombotik mikroanjiyopati (TMA) olaylar�, hastalar�n %1'inden az (3/373) ve HEMLIBRA ile tedavi edilirken en az bir doz aPCC alm�� hastalar�n %9,7'sinde (3/31) bildirilmi�tir. �� TMA'n�n t�m�, 24 saat veya daha uzun s�re
>100 U/Kg/24 saat ortalama k�m�latif miktarda aPCC uyguland���nda meydana gelmi�tir (bkz. B�l�m 4.4). Hastalar trombositopeni, mikroanjiyopatik hemolitik anemi ve ADAMTS13 aktivitesinde �iddetli eksiklik olmaks�z�n akut b�brek hasar� ile ba�vurmu�tur. Bir hasta, TMA olaylar�n�n tekrarlamadan d�zelmesini takiben HEMLIBRA kullan�m�na devam etmi�tir.
Trombotik olaylar
Havuzlanm�� faz III klinik �al��malar�nda ciddi trombotik olaylar hastalar�n %1'inden az (2/373) ve HEMLIBRA ile tedavi edilirken en az bir aPCC dozu alm�� hastalar�n %6,5'inde (2/31) bildirilmi�tir. Her iki ciddi trombotik olay da 24 saat veya daha uzun s�re >100 U/Kg/24 saat ortalama k�m�latif miktarda aPCC uyguland���nda meydana gelmi�tir. Bir hasta, trombotik olaylar�n tekrarlamadan d�zelmesini takiben HEMLIBRA kullan�m�na devam etmi�tir (bkz. b�l�m 4.4).
Pivotal klinik �al��malarda HEMLIBRA ve aPCC tedavisi aras�ndaki etkile�imin karakterizasyonu
HEMLIBRA profilaksisi alan hastalarda 82 aPCC tedavisi* uygulanm�� olup, bunlar�n sekizinde (%10) 24 saat veya daha uzun s�re >100 U/Kg/24 saat ortalama k�m�latif miktarda aPCC uygulanm��t�r, bu sekiz �rne�in ikisi trombotik olaylar ve sekizden ��� TMA ile ili�kilidir (Tablo 3). Geri kalan aPCC tedavileri ile ili�kili bir TMA veya trombotik olay s�z konusu olmam��t�r. T�m aPCC tedavilerinden %68'i <100 U/kg'l�k tek bir inf�zyondan ibarettir.
Tablo 3 Havuzlanm�� faz III klinik �al��malar�nda aPCC tedavisinin* karakterizasyonu
aPCC tedavisinin s�resi | 24 saatte ortalama k�m�latif aPCC miktar� (U/kg/24 saat) | ||
<50 | 50-100 | >100 | |
<24 saat | 9 | 47 | 13 |
24-48 saat | 0 | 3 | 1 |
>48 saat | 1 | 1 | 7 |
Enjeksiyon yeri reaksiyonlar�
Enjeksiyon yeri reaksiyonlar� (EYR'ler) klinik �al��malarda �ok yayg�n (%20) olarak bildirilmi�tir. HEMLIBRA klinik �al��malar�nda g�zlenen t�m EYR'ler, ciddi olmayan olaylar olarak bildirilmi�tir ve hafif ila orta �iddettedirler. EYR'lerin %95'i tedavi uygulanmadan ortadan kalkm��t�r. En yayg�n �ekilde bildirilen EYR semptomlar�, enjeksiyon yerinde k�zar�kl�k (%11), enjeksiyon yerinde a�r� (%4) ve enjeksiyon yerinde ka��nt� (pruritus) (%3) olmu�tur.
�mm�nojenisite
Hemlibra ile havuzlanm�� Faz III klinik �al��malarda, emicizumab konsantrasyonunun azalmas�yla ili�kili n�tralize edici anti-emicizumab antikorlar�n�n geli�imi yayg�n de�ildir (bkz. B�l�m 5.1). Emicizumab konsantrasyonunun azalmas�yla n�tralize edici anti- emicizumab antikorlar� geli�tiren bir hasta, be� haftal�k tedaviden sonra etkililik kayb� ya�am��t�r (ara kanama olarak kendini g�sterdi) ve daha sonra Hemlibra tedavisini b�rakm��t�r (bkz. B�l�m 4.4 ve 5.1).
Pediyatrik pop�lasyon:
Ara�t�r�lan pediyatrik pop�lasyon, 5'i (%5) bebek (1 ay ila 2 ya� alt�), 55'i (%51) �ocuk (2 ya� sonras� ila 12 ya� alt�) ve 47'si (%44) ergen (12 ya� ila 18 ya� alt�) olan toplamda 107 hastadan olu�ur.
HEMLIBRA'n�n g�venlilik profili bebekler, �ocuklar, ergenler ve yeti�kinler aras�nda genel olarak tutarl� bulunmu�tur.
��pheli advers reaksiyonlar�n raporlanmas�
Ruhsatland�rma sonras� ��pheli ila� advers reaksiyonlar�n�n raporlanmas� b�y�k �nem ta��maktad�r. Raporlama yap�lmas�, ilac�n yarar/risk dengesinin s�rekli olarak izlenmesine olanak sa�lar. Sa�l�k mesle�i mensuplar�n�n herhangi bir ��pheli advers reaksiyonu T�rkiye Farmakovijilans Merkezi (T�FAM)'ne bildirmeleri gerekmektedir (www.titck.gov.tr; e- posta: tufam@titck.gov.tr; tel: 0 800 314 00 08; faks: 0312 218 35 99).
4.9. Doz a��m� ve tedavisi
HEMLIBRA doz a��m� ile ilgili deneyim k�s�tl�d�r. Semptomlar
Kazara doz a��m� hiperkoag�labiliteye yol a�abilir. Tedavi
Kazara doz a��m�n�n oldu�u hastalar acilen hekimleriyle ileti�ime ge�meli ve yak�ndan izlenmelidir.
5. FARMAKOLOJ�K �ZELL�KLER
5.1. Farmakodinamik �zellikler
Farmakoterap�tik grup: Antihemorajikler, di�er sistemik hemostatikler ATC kodu: B02BX06
Etki mekanizmas�
Emicizumab, bispesifik antikor yap�s� olan h�manize monoklonal modifiye edilmi� imm�noglob�lin G4 (IgG4) antikorudur.
Emicizumab, etkili hemostaz i�in gerekli olan eksik aktive fakt�r VIII'in fonksiyonunun yerine konmas� i�in aktive fakt�r IX ve fakt�r X'u birle�tirir.
Emicizumab�n fakt�r VIII ile yap�sal ili�kisi veya sekans homolojisi yoktur ve bu haliyle fakt�r VIII'e kar�� direkt antikor geli�imini ind�klemez ya da artt�rmaz.
Farmakodinamik
HEMLIBRA ile profilaksi tedavisi, aPTT'yi k�salt�r ve testlerde bildirilen fakt�r VIII aktivitesini (insan koag�lasyon fakt�rleri ile bir kromojenik tayin kullan�larak) artt�r�r. Bu iki farmakodinamik belirte� emicizumab�n ger�ek in vivo hemostatik etkisini yans�tmamaktad�r (aPTT a��r� derecede k�salm��t�r ve bildirilen fakt�r VIII aktivitesi oldu�undan daha y�ksek ��kabilir); ancak emicizumab�n prokoag�lan etkisi ile ilgili ba��l bir g�sterge sa�lamaktad�r.
Klinik etkililik ve g�venlilik
HEMLIBRA'n�n FVIII inhibit�rl� veya inhibit�rs�z hemofili A hastalar�n�n rutin profilaksisinde etkilili�i d�rt klinik �al��mada (�� eri�kin ve ergen �al��mas� [HAVEN 3, HAVEN 1 ve HAVEN 4] ve bir pediyatrik �al��mada [HAVEN 2]) de�erlendirilmi�tir.
Ad�lesan ve eri�kinlerde yap�lan klinik �al��malar
FVIII inhibit�rs�z hemofili A hastalar� (≥ 12 ya� ve > 40 kg) (�al��ma BH30071 – HAVEN 3)
HAVEN 3 �al��mas�, daha �nce FVIII ile kanad�k�a (“ihtiya� halinde”) veya profilaksi tedavi alm�� olan, FVIII inhibit�rs�z hemofili A hastas� 152 ad�lesan ve eri�kin erkekte (> 12 ya� ve
> 40 kg) yap�lm�� olan randomize, �ok merkezli, a��k etiketli bir faz III klinik �al��mad�r. Hastalar ilk d�rt hafta boyunca haftada bir kez 3 mg/kg ve bunu takiben haftada bir kez 1,5 mg/kg (Kol A ve D) veya iki haftada bir kez 3 mg/kg (Kol B) subkutan HEMLIBRA alm�� veya profilaksi almam��t�r (Kol C). Kol C'deki hastalar profilaksi uygulanmaks�z�n en az 24 haftay� tamamlad�ktan sonra HEMLIBRA'ya (iki haftada bir 3 mg/kg) ge�ebilmi�tir. Kol A ve B'de, iki veya daha fazla anlaml� kanama (kararl� durumda iken meydana gelen spontan ve klinik olarak anlaml� kanamalar) ge�iren hastalar i�in haftada bir kez 3 mg/kg doz titrasyonuna izin verilmi�tir. Kol D hastalar�nda, ikinci anlaml� kanamadan sonra doz titrasyonu yap�labilmi�tir. Ara analiz yap�ld��� s�rada, be� hastan�n idame dozunda yukar� titrasyonun yap�lm�� oldu�u saptanm��t�r.
Daha �nce kanad�k�a (“ihtiya� halinde”) FVIII ile tedavi edilmi� olan 89 hasta, haftada bir kez HEMLIBRA almak �zere (Kol A; N = 36), iki haftada bir kez HEMLIBRA almak �zere (Kol B; N = 35) veya hi� profilaksi almamak �zere (Kol C; N = 18) 2:2:1 oranda randomize edilmi�, �nceki 24 haftal�k kanama oran�na g�re (< 9 veya ≥ 9) katmanlama yap�lm��t�r. Daha �nce FVIII profilaksisi ile tedavi edilmi� olan 63 hasta, HEMLIBRA (haftada bir kez 1,5 mg/kg) almak �zere Kol D'ye kaydedilmi�tir.
�al��man�n birincil amac�, daha �nce kanad�k�a FVIII ile tedavi edilmi� olan hastalarda haftada bir kez (Kol A) veya iki haftada bir kez (Kol B) uygulanan HEMLIBRA profilaksisinin, hi� profilaksi uygulanmamas�na (Kol C) k�yasla etkilili�ini, koag�lasyon fakt�rleri ile tedavi gerektiren kanamalar�n say�s�na dayanarak de�erlendirmek olmu�tur (bkz. Tablo 4). �al��man�n di�er ama�lar�, Kol A veya B ile Kol C'nin randomize kar��la�t�rmas�n�n HEMLIBRA profilaksisinin t�m kanamalar�n, spontan kanamalar�n, eklem kanamalar�n�n ve hedef eklem kanamalar�n�n say�s�n� azaltmadaki etkilili�i a��s�ndan de�erlendirilmesini (bkz. Tablo 4) ve hasta bildirimli sa�l�k ile ilgili ya�am kalitesinin (HRQoL) �l��lmesini i�ermi�tir. Ayr�ca, bir tercih anketi kullan�larak hastalar�n tedavi tercihi de de�erlendirilmi�tir.
HEMLIBRA profilaksisinin etkilili�i, �al��maya kaydedilmeden �nce giri�imsel olmayan bir �al��maya kat�lm�� olan hastalarda �nceki profilaktik FVIII tedavisi (Kol D) ile de kar��la�t�r�lm��t�r (bkz. Tablo 5). Bu kar��la�t�rmaya yaln�zca giri�imsel olmayan �al��madan gelen hastalar dahil edilmi�tir; ��nk� kanama ve tedavi verileri HAVEN 3'teki ile ayn� veri derinli�i d�zeyinde toplanm��t�r.
Giri�imsel olmayan �al��ma g�zlemsel bir �al��ma olup, temel amac� hemofili A hastalar�nda giri�imsel �al��ma d�zeni d���nda kanama epizotlar� ve hemofili ila�lar�n�n kullan�m� hakk�nda detayl� klinik verilerin yakalanmas� olmu�tur.
Fakt�r VIII inhibit�rl� (>12 ya�) hemofili A hastalar� (�al��ma BH29884 – HAVEN 1)
HAVEN 1 �al��mas�, daha �nce bypass edici ajanlar (aPCC ve rFVIIa) ile kanad�k�a tedavi ya da profilaksi almakta olan, fakt�r VIII inhibit�rl� hemofili A hastas� 109 ad�lesan ve eri�kin erkek (ya�lar� ≥ 12) �zerinde ger�ekle�tirilen bir randomize, �ok merkezli, a��k etiketli klinik �al��mad�r. �al��mada, hastalara haftal�k HEMLIBRA profilaksisi (Grup A, C ve D) - 4 hafta boyunca haftada bir kez 3 mg/kg, ard�ndan haftada bir kez 1,5 mg/kg - ya da kanad�k�a tedavi uygulanm��t�r (Grup B). Grup B'deki randomize hastalar, profilaksi olmadan en az 24 haftay� tamamlad�ktan sonra HEMLIBRA profilaksisine ge�ebilmi�tir. 2 ya da daha fazla anlaml� kanama ge�irmi� hastalarda (kararl� halde spontan ve klinik olarak kan�tlanm�� anlaml� kanaman�n olmas� durumunda) HEMLIBRA profilaksisinde 24 hafta ge�irildikten sonra haftada bir kez 3 mg/kg'a ��kar�lacak �ekilde doz titrasyonuna izin verilmi�tir. Primer analiz s�resi boyunca, iki hastan�n idame dozlar� haftada bir kez 3 mg/kg'a y�kseltilmi�tir.
Daha �nce bypass edici ajanlarla kanad�k�a tedavi almakta olan 53 hasta, �al��madan �nceki
24 haftal�k kanama say�lar�na (<9 veya ≥ 9) g�re yap�lan s�n�fland�rmayla birlikte HEMLIBRA profilaksisi (Grup A) ya da kanad�k�a tedavi alacak (Grup B) �ekilde 2:1 oran�nda randomize edilmi�lerdir.
Daha �nce bypass edici ajan profilaksisi ile tedavi edilen 49 hasta, HEMLIBRA profilaksisi almak i�in Grup C'ye kaydedilmi�tir. Kay�ttan �nce NIS'e (Giri�imsel olmayan �al��ma) kat�lan, ancak A ve B gruplar�n�n kapat�lmas�ndan �nce HAVEN 1'e kaydolamam��, kanad�k�a bypass edici ajan tedavisi almakta olan 7 hasta, HEMLIBRA profilaksisi almak i�in Grup D'ye kaydedilmi�lerdir.
�al��man�n temel amac�, �ncesinde kanad�k�a bypass edici ajan tedavisi almakta olan hastalarda, kanad�k�a tedavi ile kar��la�t�rmal� olarak, haftal�k HEMLIBRA profilaksisinin, zaman i�inde (en az 24 hafta veya kesilme tarihi) (Bkz. Tablo 6), koag�lasyon fakt�rleri ile tedaviyi gerektiren kanama say�s� �zerindeki etkisini de�erlendirmektir (Grup A'ya kar��l�k Grup B). A ve B gruplar�n�n randomize olarak kar��la�t�r�lmas�n�n di�er ikincil hedefleri, t�m kanamalar�n, spontan kanamalar�n, eklem kanamalar�n�n ve hedef eklem kanamalar�n�n (Bkz. Tablo 6) say�s�n�n azalt�lmas�nda HEMLIBRA profilaksisinin etkilili�ini ve bunun yan� s�ra hastalar�n sa�l�kla ili�kili ya�am kaliteleri ile sa�l�k durumlar�n� de�erlendirmektir (Bkz. Tablo 9 ve 10). �al��madaki t�m hastalar i�in ortalama maruziyet s�resi (+SD) 21,38 haftad�r (12,01). Her bir tedavi kolu i�in ortalama maruziyet s�releri (+SD) Kol A i�in 28,86 hafta (8,37), Kol B i�in 8,79 (3,62), Kol C i�in 21,56 (11,85) ve Kol D i�in 7,08 haftad�r (3,89). Kol A'daki bir hasta HEMLIBRA ba�lat�lmadan �al��madan �ekilmi�tir.
�al��ma ayn� zamanda, kay�t �ncesi NIS'e kat�lan hastalarda (s�ras�yla Grup A ve C) (Bkz. Tablo 7) daha �nce uygulanm�� olan bypass edici ajanlar ile kanad�k�a tedavi ya da profilaksi kar��s�nda (ayr� kar��la�t�rmalar) haftal�k HEMLIBRA profilaksisinin etkilili�ini de de�erlendirmi�tir.
Fakt�r VIII inhibit�rl� ya da inhibit�rs�z hemofili A hastalar� (≥ 12 ya�) (�al��ma BO39182 – HAVEN 4)
HEMLIBRA, daha �nce bypass edici ajanlar� veya FVIII ile kanad�k�a (“ihtiya� halinde”) ya da profilaksi alm�� olan, FVIII inhibit�rl� veya inhibit�rs�z 41 eri�kin ve ad�lesan hemofili A hastas� erkekte (≥ 12 ya� ve > 40 kg) yap�lan tek kollu, �ok merkezli bir faz III klinik �al��mada ara�t�r�lm��t�r. Hastalar, d�rt hafta boyunca haftada bir kez 3 mg/kg ve bunu takiben d�rt haftada bir 6 mg/kg HEMLIBRA profilaksisi alm��lard�r.
�al��man�n birincil amac�, d�rt haftada bir verilen HEMLIBRA profilaksisinin yeterli kanama kontrol�n�n s�rd�r�lmesindeki etkilili�ini, tedavi edilen kanamalara dayanarak de�erlendirmek olmu�tur. Di�er ama�lar, HEMLIBRA profilaksisinin t�m kanamalar, tedavi edilen spontan kanamalar, tedavi edilen eklem kanamalar� ve tedavi edilen hedef eklem kanamalar� �zerindeki klinik etkilili�ini de�erlendirmek olmu�tur (bkz. Tablo 8). Ayr�ca, bir tercih anketi kullan�larak hastalar�n tedavi tercihi de de�erlendirilmi�tir.
Eri�kinlerde ve Ad�lesanlarda Etkililik Bulgular� HAVEN 3
T�m kanamalar, tedavi edilen spontan kanamalar, tedavi edilen eklem kanamalar� ve tedavi edilen hedef eklem kanamalar� i�in kanama oran� a��s�ndan HEMLIBRA profilaksisinin hi� profilaksi uygulanmamas�na k�yasla etkililik bulgular� Tablo 4'te g�sterilmektedir.
Tablo 4 HAVEN 3 �al��mas�: ≥12 ya� inhibit�rs�z hastalarda profilaksi uygulanmamas�na k�yasla HEMLIBRA profilaksisi ile y�ll�k kanama say�lar�
Sonlan�m noktas� | C kolu: Profilaksi yok (N=18) | A kolu: Haftal�k 1,5 mg/kg HEMLIBRA (N=36) | B kolu: 2 haftada 1 kez 3 mg/kg HEMLIBRA (N=35) |
Tedavi edilen kanamalar | |||
Y�ll�k kanama say�s� (ABR) (%95 GA) | 38,2(22,9; 63,8) | 1,5 (0,9; 2,5) | 1,3 (0,8; 2,3) |
% azalma (RR), p-de�eri | Uygulanabilir de�il | %96 (0,04), <0,0001 | %97 (0,03), <0,0001 |
0 kanama izlenen hasta y�zdesi (%95 GA) | 0 (0; 18,5) | 55,6 (38,1; 72,1) | 60 (42,1; 76,1) |
Medyan y�ll�k kanama say�s� (ABR) (IQR) | 40,4 (25,3; 56,7) | 0 (0; 2,5) | 0 (0; 1,9) |
T�m kanamalar | |||
Y�ll�k kanama say�s� (ABR) (%95 GA) | 47,6 (28,5; 79,6) | 2,5 (1,6; 3,9) | 2,5 (1,6; 4,3) |
% azalma (RR), p-de�eri | Uygulanabilir de�il | %95 (0,05), <0,0001 | %94 (0,06), <0,0001 |
0 kanama izlenen hasta y�zdesi (%95 GA) | 0 (0; 18,5) | 50 (32,9; 67,1) | 40 (23,9; 57,9) |
Tedavi edilen spontan kanamalar | |||
Y�ll�k kanama say�s� (ABR) (%95 GA) | 15,6 (7,6; 31,9) | 1 (0,5; 1,9) | 0,3 (0,1; 0,8) |
Uygulanabilirde�il | |||
0 kanama izlenen hasta y�zdesi (%95 GA) | 22,2 (6,4; 47,6) | 66,7 (49; 81,4) | 88,6 (73,3; 96,8) |
Tedavi edilen eklem kanamalar� | |||
Y�ll�k kanama oran� (ABR) (%95 GA) | 26,5 (14,67; 47,79) | 1,1 (0,59; 1,89) | 0,9 (0,44; 1,67) |
% azalma (RR), p-de�eri | Uygulanabilir de�il | %96 (0,04), <0,0001 | %97 (0,03), <0,0001 |
0 kanama izlenen hasta y�zdesi (%95 GA) | 0 (0; 18,5) | 58,3 (40,8; 74,5) | 74,3 (56,7; 87,5) |
Tedavi edilen hedef eklem kanamalar� | |||
Y�ll�k kanama say�s� (ABR) (%95 GA) | 13 (5,2; 32,3) | 0,6 (0,3; 1,4) | 0,7 (0,3; 1,6) |
% azalma (RR), p-de�eri | Uygulanabilir de�il | %95 (0,05), <0,0001 | %95 (0,05), <0,0001 |
0 kanama izlenen hasta y�zdesi (%95 GA) | 27,8 (9,7; 53,5) | 69,4 (51,9; 83,7) | 77,1 (59,9; 89,6) |
HAVEN 3 klinik �al��mas�nda ger�ekle�tirilen hasta i�i analizlerinde, hastalar �al��maya dahil edilmeden �nce giri�imsel olmayan �al��madan toplanan FVIII profilaksisi verileri ile k�yasland���nda, HEMLIBRA profilaksisini tedavi edilen kanama say�s�nda istatistiksel olarak anlaml� (p<0,0001) bir azalma sa�lam��t�r (%68) (bkz. Tablo 5).
Tablo 5 HAVEN 3 �al��mas�: FVIII profilaksisine k�yasla HEMLIBRA profilaksisi ile y�ll�k kanama say�lar�na ait hasta i�i de�erlendirmesi
Sonlan�m noktas� | D NIS kolu: �nceki FVIII profilaksisi (N=48) | D kolu: Haftal�k 1,5 mg/kg HEMLIBRA (N=48) |
Medyan Etkililik S�resi (hafta) | 30,1 | 33,7 |
Tedavi edilen kanamalar | ||
Y�ll�k kanama say�s� (ABR) (%95 GA) | 4,8 (3,2; 7,1) | 1,5 (1; 2,3) |
% azalma (%95 GA), p- de�eri | % 68 (% 48,6; %80,5), <0,0001 | |
0 kanama izlenen hasta y�zdesi (%95 GA) | 39,6 (25,8; 54,7) | 54,2 (39,2; 68,6) |
Medyan y�ll�k kanama say�s� (ABR) (IQR) | 1,8 (0; 7,6) | 0 (0; 2,1) |
HAVEN 1
Profilaksi almayan hastalar ile k�yasland���nda HEMLIBRA profilaksisinin tedavi edilen kanamalar, t�m kanamalar, tedavi edilen spontan kanamalar, tedavi edilen eklem kanamalar� ve tedavi edilen hedef eklem kanamalar� i�in oranlar a��s�ndan etkililik sonu�lar� Tablo 6'da g�sterilmektedir.
Tablo 6 HAVEN 1 �al��mas�: ≥12 ya� FVIII inhibit�rl� hastalarda profilaksi uygulanmamas�na k�yasla HEMLIBRA profilaksisi ile y�ll�k kanama say�lar�
Sonlan�m Noktas� | Grup B: profilaksi yok | Grup A: Haftal�k 1,5 mg/kg HEMLIBRA |
| N=18 | N=35 |
Tedavi Edilen Kanamalar |
| |
ABR (%95 GA) | 23,3 (12,33; 43,89) | 2,9 (1,69; 5,02) |
% azalma (%95 GA), p-de�eri | %87 (0,13), < 0,0001 | |
0 kanama izlenen hasta %'si (%95 GA) | 5,6 (0,1; 27,3) | 62,9 (44,9; 78,5) |
Medyan ABR (IQR) | 18,8 (12,97; 35,08) | 0 (0; 3,73) |
T�m Kanamalar | ||
ABR (%95 GA) | 28,3 (16,79; 47,76) | 5,5 (3,58; 8,60) |
% azalma (%95 GA), p-de�eri | %80 (0,20), < 0,0001 | |
0 kanaman�n oldu�u izlenen hasta %'si (%95 GA) | 5,6 (0,1; 27,3) | 37,1 (21,5; 55,1) |
Tedavi Edilen Spontan Kanamalar |
| |
ABR (%95 GA) | 16,8 (9,94; 28,30) | 1,3 (0,73; 2,19) |
% azalma (%95 GA), p-de�eri | %92 (%0,08), < 0,0001 | |
0 kanaman�n oldu�u izlenen hasta %'si (%95 GA) | 11,1 (1,4; 34,7) | 68,6 (50,7; 83,1) |
Tedavi Edilen Eklem Kanamalar� |
| |
ABR (%95 GA) | 6,7 (21,99; 22,42) | 0,8 (0,26; 2,20) |
% azalma (%95 GA), p-de�eri | %89 (%0,11), 0,0050 | |
0 kanaman�n oldu�u izlenen hasta %'si (%95 GA) | 50 (26; 74) | 85,7 (69,7; 95,2) |
Tedavi Edilen HedefEklem | ||
Kanamalar� |
| |
ABR (%95 GA) | 3 (0,96; 9,13) | 0,1 (0,03; 0,58) |
% azalma (%95 GA), p-de�eri | %95 (%0,05), 0,0002 | |
0 kanama izlenen hasta %'si (%95 GA) | 50 (26; 74) | 94,3 (80,8; 99,3) |
G�ven aral��� (GA), negatif binomiyal regresyon (NBR) modelinden ve p-de�eri Katmanla�t�r�lm�� Wald testinden al�nm�� olup, belirtilmi� kollar aras�nda ABR'yi kar��la�t�rmaktad�r. Grup B: yaln�zca profilaksi uygulanmayan d�nemi kapsar. Kanama tan�mlar� ISTH kriterlerine dayal�d�r. Tedavi edilmi� kanamalar: bypass edici ajanlar ile tedavi edilmi� kanamalar. T�m kanamalar: bypass edici ajanlar ile tedavi edilmi� ve tedavi edilmemi� kanamalar Dozu yukar� titre edilmi� hastalar i�in yaln�zca yukar� titrasyondan �nceki verileri i�erir. Emicizumaba maruz kalm�� hastalar, tedaviye 4 hafta boyunca haftada 1 kez 3 mg/kg/g�n y�kleme dozuyla ba�lam��lard�r. ABR = Y�ll�k Kanama Say�s�; GA = g�ven aral���; IQR = �eyrekler a��kl���, 25. ile 75. y�zdelik dilim aras� | ||
HAVEN 1 hasta i�i analizinde, �ncesinde giri�imsel olmayan �al��mada bypass edici ajan profilaksisi ile elde edilen sonu�lara k�yasla, HEMLIBRA profilaksisi ile tedavi edilen kanama say�lar�nda istatistik (p=0,003) ve klinik olarak anlaml� (%79) azalma ortaya ��km��t�r (bkz. Tablo 7).
Tablo 7 HAVEN 1: HEMLIBRA Profilaksisine kar�� �ncesinde bypass edici ajanla profilaksi alm�� hastalar�n (NIS Hastalar�) Y�ll�k Kanama Say�s� i�in kendi i�erisinde kar��la�t�rmas�
Sonlan�m Noktas� | Cgrubu: Daha �nce bypass edici ajan profilaksisi | Grup C: haftada bir 1,5 mg/kg HEMLIBRA |
| N = 24 | N = 24 |
Tedavi Edilen Kanamalar | ||
ABR (%95 GA) | 15,7 (11,08; 22,29) | 3,3 (1,33; 8,08) |
0 kanama izlenen hasta y�zdesi (%95 GA) | 12,5 (2,7; 32,4) | 70,8 (48,9; 87,4) |
Ortalama ABR (IQR) | 12 (5,73; 24,22) | 0 (0; 2,23) |
% azalma (RR), p-de�eri | %79 (0,21); 0,0003 | |
| ||
HAVEN 4
4 haftada 1 uygulanan HEMLIBRA profilaksisinin tedavi edilen kanamalar, t�m kanamalar, tedavi edilen spontan kanamalar, tedavi edilen eklem kanamalar� ve tedavi edilen hedef eklem kanamalar� a��s�ndan etkililik sonu�lar� Tablo 8'de g�sterilmektedir. ≥12 ya��ndaki 41 hasta, medyan 25,6 hafta s�resince de�erlendirilmi�tir (24,1-29,4 aral���).
Tablo 8 HAVEN 4: ≥12 ya� Fakt�r VIII inhibit�rl� ve inhibit�rs�z hastalarda HEMLIBRA profilaksisi ile y�ll�k kanama say�lar�
| 4 haftada 1 kez 6 mg/kg HEMLIBRA | ||
Sonlan�m noktalar� | 0 kanama y�zdesi (%95 GA) | ||
N | 41 | 41 | 41 |
Tedavi edilen kanamalar | 2,4 (1,4; 4,3) | 0 (0; 2,1) | 56,1 (39,7; 71,5) |
T�m kanamalar | 4,5 (3,1; 6,6) | 2,1 (0; 5,9) | 29,3 (16,1; 45,5) |
Tedavi edilen spontan kanamalar | 0,6 (0,3; 1,5) | 0 (0; 0) | 82,9 (67,9; 92,8) |
Tedavi edilen eklem kanamalar� | 1,7 (0,8; 3,7) | 0 (0; 1,9) | 70,7 (54,5; 83,9) |
Tedavi edilen hedef eklem kanamalar� | 1 (0,3; 3,3) | 0 (0; 0) | 85,4 (70,8; 94,4) |
Eri�kin ve Ad�lesanlarda Sa�l�k �le �lgili Sonu� �l��tleri
Eri�kin ve ad�lesanlar�n dahil oldu�u HAVEN klinik �al��malar�nda, hasta bildirimli sonu�lar birka� �l��t ile de�erlendirilmi�tir. Hastalarda hemofili ile ilgili ya�am kalitesi, eri�kinler (>18 ya�) i�in Hemofiliye �zg� Ya�am Kalitesi (Haem-A-QoL) anketi ve bunun ergen versiyonu (Haemo-QoL-SF, 8 ila <18 ya� i�in) ile de�erlendirilmi�tir. Haem-A-QoL ve Haemo-QoL-SF i�in, Fiziksel Sa�l�k Skoru (yani a�r�l� �i�likler, eklem a�r�s� varl��� hareket ile a�r�, uza�a y�r�mede g��l�k ve haz�rlanmak i�in gereken s�renin artmas�) ve Toplam Skor (t�m skorlar�n �zeti) protokolde tan�mlanm�� ilgi konusu sonlan�m noktalar�d�r. Sa�l�k durumundaki de�i�imi �l�mek i�in, EuroQoL Be� Boyutlu Be� D�zeyli Anketine (EQ-5D- 5L) ait G�rsel Analog Skala (VAS) ve �ndeks Yararl�l�k Skoru (IUS) incelenmi�tir.
HAVEN 1 Sa�l�kla �li�kili Sonu�lar
Bu �al��mada, ≥18 ya� hastalar i�in sa�l�kla ili�kili sonu�lar, 25. haftada eri�kinlere y�nelik (Bkz. Tablo 9) Haem-A-QoLanketiilede�erlendirilmi�tir. Ba�lang�� Toplam Skorlar�
(ortalama = s�ras�yla 41,14 ve 44,58) ve Fiziksel Sa�l�k �l�e�i skorlar� (ortalama = s�ras�yla 52,41 ve 57,19) HEMLIBRA profilaksisi ve kanad�k�a tedavi grubu i�in benzerdir. Tablo 9'da HEMLIBRA profilaksisi kolu (Kol A) ve profilaksi almayan kol (Kol B) aras�nda 24 haftal�k tedavi sonras� Haem-A-QoL Toplam Skoru ve Fiziksel Sa�l�k �l�e�ine ili�kin kar��la�t�rman�n bir �zeti sunulmaktad�r. Haftal�k HEMLIBRA profilaksisi �nceden belirlenmi� sonlan�m noktalar� olan 25. Hafta de�erlendirmesinde Haem-A-QoL Toplam Skoru ve Fiziksel Sa�l�k �l�e�i skorunda kanad�k�a tedaviye k�yasla istatistiksel ve klinik anlaml� bir iyile�me g�stermi�tir.
Tablo 9 HAVEN 1: ≥18 ya� Fakt�r VIII inhibit�rl� hastalarda profilaksi uygulanmas�na k�yasla HEMLIBRA profilaksisi ileHaem-A-QoL skorlar�ndaki de�i�im
25. haftada Haem-A-QoL Skorlar� | Grup B: profilaksi yok (N=14) | Grup A: 1,5 mg/kg Haftal�k HEMLIBRA (N=25) |
Toplam Skor (0 ila 100 aral���) | ||
D�zeltilmi� ortalama | 54,17 | 32,61 |
D�zeltilmi� ortalamalarda farkl�l�k (%95 GA) | 21,55 (7,89; 35,22) | |
p-de�eri | 0,0029 | |
Fiziksel Sa�l�k skoru (0 ila 100 aral���) | ||
D�zeltilmi� ortalama | 43,21 | 29,2 |
D�zeltilmi� ortalamalarda farkl�l�k (%95 GA) | 14,01 (5,56; 22,45) | |
Grup B: yaln�zca profilaksi uygulanmayan d�nemi kapsar. Dozu yukar� titre edilmi� hastalar i�in yaln�zca yukar� titrasyondan �nceki verileri i�erir. Emicizumaba maruz kalm�� hastalar, tedaviye 4 hafta boyunca haftada 1 kez 3 mg/kg/g�n y�kleme dozuyla ba�lam��lard�r. Haem-A_QoL �l�ekleri 0 ila 100 aras�nda de�i�ir, d���k skorlar daha iyi HRQoL'ye i�aret etmektedir. Klinik olarak anlaml� farkl�l�k: Toplam skor: 7 puan; Fiziksel Sa�l�k: 10 puan. Analizler hem ba�lang�� noktas�nda, hem de 25.Hafta de�erlendirmesinde yan�t veren bireylerden elde edilen verilere dayanmaktad�r. | ||
HAVEN 1 Sa�l�k Durumu Sonu�lar�
Tablo 10'da HEMLIBRA profilaksisi kolu (Kol A) ve profilaksi almayan kol (Kol B) aras�nda 24 haftal�k tedavi sonras� EQ-5D-5L indeksi fayda �l�e�i ve g�rsel analog �l�e�e ili�kin kar��la�t�rman�n bir �zeti sunulmaktad�r.
![]()
Tablo 10 HAVEN 1 : 25. haftada ≥12 ya��ndaki hastalarda EQ-5D-5L skorlar�
24 haftadan sonra EQ-5D- 5L | Grup B: profilaksi yok (N=16) | Grup A: 1,5 mg/kg Haftal�k HEMLIBRA (N=29) |
G�rsel Analog �l�ek |
| |
D�zeltilmi� ortalama | 74,36 | 84,08 |
D�zeltilmi� ortalamalarda farkl�l�k (%95 GA) | -9,72 (-17,62; -1,82) | |
�ndeks Fayda Skoru |
| |
0,81 | ||
D�zeltilmi� ortalamalarda farkl�l�k (%95 GA) | -0,16 (-0,25; -0,07) |
Grup B: yaln�zca profilaksi uygulanmayan d�nemi kapsar. Dozu yukar� titre edilmi� hastalar i�in yaln�zca yukar� titrasyondan �nceki verileri i�erir. Emicizumaba maruz kalm�� hastalar, tedaviye 4 hafta boyunca haftada 1 kez 3 mg/kg/g�n y�kleme dozuyla ba�lam��lard�r. Daha y�ksek skorlar daha iyi ya�am kalitesine i�aret etmektedir. Klinik a��dan �nemli farkl�l�k: VAS: 7 puan; �ndeks fayda skoru: 0,07 puan Analizler hem ba�lang�� noktas�nda, hem de 25.Hafta de�erlendirmesinde yan�t veren bireylerden elde edilen verilere dayanmaktad�r. | |
Pediyatrik hastalarda klinik �al��ma
Fakt�r VIII inhibit�rl� pediyatrik hemofili A hastalar� (ya� <12 ya� veya <40 kg a��rl���nda 12 ila 17 ya�) (�al��ma BH29992 – HAVEN 2)
Haftal�k HEMLIBRA profilaksisi, fakt�r VIII inhibit�rl� pediyatrik hemofili A hastalar�nda (12 ya� alt� veya 12 ila 17 ya� aras�nda, <40 kg a��rl���nda), tek kollu, �ok merkezli, a��k etiketli bir klinik �al��mada de�erlendirilmi�tir. Hastalar, ilk 4 hafta boyunca haftada bir kez 3 mg/kg ve daha sonra haftada bir kez 1,5 mg/kg olacak �ekilde HEMLIBRA profilaksisi alm��t�r.
�al��mada, kay�ttan �nce NIS'e kat�lan hastalarda (kendi i�erisinde de�erlendirilen hastalar) daha �nceki kanad�k�a ya da profilaktik bypass edici ajan tedavisi ile kar��la�t�rmal� olarak, haftal�k HEMLIBRA profilaksisinin etkilili�i farmakokinetik �zellikleri ve g�venlili�i de�erlendirmi�tir.
HAVEN 2 �al��mas�n�n Pediyatrik Etkililik Bulgular� (Ara Analiz)
Ara analizler s�ras�nda < 2 ya��nda olan 4 hasta, 2 ila < 6 ya� aras�nda olan 17 hasta ve 6 ila < 12 ya� aras�nda olan 38 hasta dahil olmak �zere, 12 ya��ndan k���k olan ve en az 12 hafta boyunca haftada 1 kez HEMLIBRA profilaksisi alan 59 hastada etkililik de�erlendirilmi�tir. 59 hasta i�in, y�ll�k olarak hesaplanm�� kanama say�s� ve s�f�r kanama g�zlenen hastalar�n y�zdesi tespit edilmi�tir (bkz. Tablo 11). Bu hastalar i�in medyan g�zlem s�resi 29,6 haftad�r (aral�k: 18,4 ila 63,0 hafta).
Tablo 11 HAVEN 2: Etkililik genel �zeti (Ara Analizler)
Sonlan�m noktas� | % S�f�r Kanama (%95 GA) | ||
Tedavi edilen kanamalar | 0,3 (0,1; 0,5) | 0 (0; 0) | 86,4 (75; 94) |
T�m kanamalar | 3,8 (2,2; 6,5) | 0 (0; 3,4) | 55,9 (42,4; 68,8 |
Tedavi edilen spontan kanamalar | 0 (0; 0,2) | 0 (0; 0) | 98,3 (90,9; 100) |
Tedavi edilen eklem kanamalar� | 0,2 (0,1; 0,4) | 0 (0; 0) | 89,8 (79,2; 96,2) |
Tedavi edilen hedef eklem kanamalar� | 0,1 (0; 0,7) | 0 (0; 0) | 96,6 (88,3; 99,6) |
Kendi i�erisinde analiz edilen hastalarda, haftal�k HEMLIBRA profilaksisi, kay�ttan �nce NIS'da toplanm�� kanama say�lar� ile kar��la�t�r�ld���nda en az 12 haftal�k tedavi alan 18 pediyatrik hastada tedavi edilmi� kanama oran�nda klinik olarak anlaml� azalma ile sonu�lanm��t�r (%98) (Tablo 12).
Tablo 12 HAVEN 2: HEMLIBRA profilaksisi uygulanan hastalar�n �nceki bypass edici ajan profilaksisine k�yasla Y�ll�k Kanama say�lar�na ait hasta i�i kar��la�t�rmas�
Sonlan�m noktas� | �nceki bypass ajan� tedavisi* (N=18) | Hemlibra profilaksisi (N = 18) |
Tedavi edilen kanamalar |
|
|
ABR (%95 GA) | 19,8 (15,3; 25,7) | 0,4 (0,15; 0,88) |
% azalma (RR) | %98 (0,02) | |
S�f�r kanama izlenen hasta y�zdesi (%95 GA) | 5,6 (0,1; 27,3) | 77,8 (52,4; 93,6) |
Medyan ABR (IQR) | 16,2 (11,49; 25,78) | 0 (0; 0) |
| ||
Pediyatrik Hastalarda Sa�l�k ile ilgili Sonu�lara Ait Bulgular HAVEN 2 �al��mas�nda Sa�l�kile�lgiliSonu�lar
HAVEN 2'de, ≥ 8 ila < 12 ya��ndaki hastalar i�in HRQoL de�erlendirmesi 25. haftada �ocuklar i�in Haemo-QoL-SF anketine dayan�larak de�erlendirilmi�tir (bkz. Tablo 13). Haemo-QoL-SF, HRQoL i�in ge�erli ve g�venilir bir �l��tt�r.
<12 ya� hastalar i�in HRQoL de�erlendirmesi de 25. haftada hasta bak�m�n� verenler taraf�ndan doldurulan Bak�mveren Y�k� Y�nlerini ��eren Adapte Edilmi� InhibQoL anketi ile ger�ekle�tirilmi�tir (bkz. Tablo 13). Adapte Edilmi� InhibQoL, HRQoL i�in ge�erli ve g�venilir bir �l��tt�r.
Tablo 13 HAVEN 2: Hasta bak�m�n� verenler taraf�ndan raporlanan HEMLIBRA profilaksisi ile hastalar�n (<12 ya�) ba�lang��tan 25. haftaya kadar Haemo- QoL-SF fiziksel sa�l�k skorlar�nda ortaya ��kan de�i�im
| Haemo-QoL-SF |
Fiziksel sa�l�k skoru (0 ila 100 aral���) | |
Ortalama ba�lang�� skoru (%95 GA) (n=18) | 29,5 (16,4 - 42,7) |
Ba�lang�ca g�re ortalama de�i�iklik (%95 GA) (n=15) | -21,7 (-37,1 - -6,3) |
| Bak�m veren Y�k� Y�nlerini ��eren Adapte Edilmi� InhibQoL |
Fiziksel sa�l�k skoru (0 ila 100 aral���) |
|
Ortalama ba�lang�� skoru (%95 GA) (n=54) | 37,2 (31,5-42,8) |
Ba�lang�ca g�re ortalama de�i�iklik (%95 GA) (n=43) | -32,4 (-38,6 – (-26,2)) |
Ameliyatlar ve giri�imler s�ras�nda bypass edici ajan veya FVIII kullan�m� konusunda deneyim s�n�rl�d�r. Ameliyatlar ve giri�imler s�ras�nda bypass edici ajan veya FVIII kullan�m� ara�t�r�c� taraf�ndan saptanm��t�r.
Ani kanama durumunda, emicizumab profilaksisi alan hastalar mevcut tedaviler ile y�netilmelidir. Bypass ajanlar� ile ilgili k�lavuz i�in bkz. B�l�m 4.4.
�mm�nojenisite
T�m terap�tik proteinlerle oldu�u gibi, HEMLIBRA ile tedavi edilen hastalarda da bir imm�n yan�t potansiyeli mevcuttur. Havuzlanm�� klinik �al��malar�nda toplam 668 hasta anti-emicizumab antikorlar�n�n varl��� a��s�ndan test edilmi�tir. Otuz d�rt hastada (%5,1) anti emicizumab antikorlar� pozitif ��km��t�r. 18 hastada (%2,7) anti-emicizumab antikorlar� in vitro olarak n�tralize edilmi�tir. Bu 18 hastadan n�tralize edici anti emicizumab antikorlar�, 14 hastada HEML�BRA'n�n farmakokineti�i veya etkinli�i �zerinde klinik olarak anlaml� bir etkiye sahip de�ilken, d�rt hastada (%0,6) emicizumab plazma konsantrasyonlar�nda d���� g�zlemlenmi�tir. N�tralize edici anti-emicizumab antikorlar� olan ve emicizumab plazma konsantrasyonlar� azalm�� bir hasta (%0,1) be� haftal�k tedaviden sonra etkililik kayb� ya�am��t�r ve HEML�BRA'y� b�rakm��t�r. Genel olarak, HEML�BRA'n�n g�venlik profili, anti-emicizumab antikorlar� olan (n�tralize edici antikorlar dahil) ve olmayan hastalar aras�nda benzerdir (bkz. B�l�m4.4ve4.8).
Etkililik kayb�na ili�kin klinik belirtiler durumunda tedavide de�i�iklik d���n�lmelidir.
Geriyatrik pop�lasyon
HEMLIBRA'n�n 65 ya� ve �st� hemofili A hastalar�nda kullan�m�, eri�kin ve ergen �al��malar� HAVEN 1, HAVEN 3 ve HAVEN 4 ile desteklenmektedir. S�n�rl� veriler temel al�nd���nda, 65 ya� veya �st� hastalarda etkililik veya g�venlilik a��s�ndan farkl�l�k oldu�una dair herhangi bir kan�t yoktur.
Pediyatrik pop�lasyon
Pediyatrik kullan�mla ilgili bilgiler i�in bkz. B�l�m 4.2.
5.2. Farmakokinetik �zellikler
Genel �zelliklerEmicizumab�n farmakokinetik �zellikleri, sa�l�kl� g�n�ll�lerde ve 389 hemofili A hastas�ndan olu�an bir veritaban�nda yap�lan bir pop�lasyon farmakokinetik analizleri kullan�larak belirlenmi�tir.
Emilim:
Hemofili A hastalar�nda subkutan uygulamay� takiben emilim yar�lanma �mr� 1,6 g�nd�r.
Hemofili A hastalar�nda ilk 4 hafta boyunca haftada bir kez 3 mg/kg'l�k subkutan uygulama sonras� emicizumab�n ortalama (± SD) �ukur plazma konsantrasyonlar� 5. haftada 52,6 ± 13,6 mcg/mL'ye ula��lm��t�r.
Tavsiye edilen idame dozlar� (haftada bir kez 1,5 mg/kg, iki haftada bir kez 3 mg/kg veya d�rt haftada bir kez 6 mg/kg) i�in kararl� durumda �ng�r�len ortalama (± SD) C, Cve C/ Coranlar� Tablo 14'de g�sterilmi�tir.
Tablo 14 Ortalama (±SD) kararl� durum emicizumab konsantrasyonlar�
| �dame dozu | ||
Parametreler | Haftada 1 kez 1,5 mg/kg | 2 haftada 1 kez 3 mg/kg | 4 haftada 1 kez 6 mg/kg |
C(mcg/mL) | 54,9±15,9 | 58,1±16,5 | 66,8±17,7 |
C(mcg/mL) | 53,5±15,7 | 53,5±15,7 | 53,5±15,7 |
C(mcg/mL) | 51,1±15,3 | 46,7±16,9 | 38,3±14,3 |
C/C | 1,08±0,03 | 1,26±0,12 | 1,85±0,46 |
Eri�kinlerde/ad�lesanlarda (≥ 12 ya�) ve �ocuklarda (< 12 ya�) haftada bir kez doz uygulamay� (4 hafta boyunca 3 mg/kg/hafta ve bunu takiben 1,5 mg/kg/hafta) takiben benzer farmakokinetik profilleri g�zlenmi�tir(bkz.�ekil1).
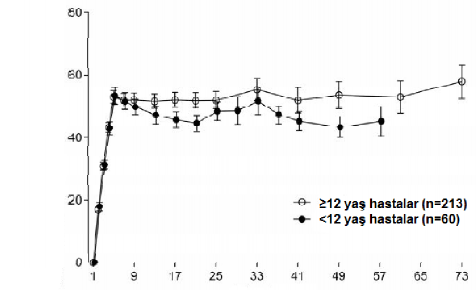
Ortalama (%95 GA) emicizumab konsantrasyonu (mcg/mL)
�ekil 1 ≥12 ya� hastalar�n ortalama plazma emcizumab konsantrasyonuna kar�� zaman profillerinin (HAVEN 1 ve HAVEN 3 �al��malar�) <12 ya� hastalar ile kar��la�t�rmas� (HAVEN 2 �al��mas�)
Zaman (Hafta)
Sa�l�kl� g�n�ll�lerde, 1 mg/kg subkutan uygulamay� takiben mutlak biyoyararlan�m, enjeksiyon b�lgesine ba�l� olarak %80,4 ile %93,1 aras�nda de�i�mi�tir. Kar�n, �st kol ve uylu�a subkutan uygulama sonras�nda benzer farmakokinetik profiller g�zlenmi�tir. Emicizumab bu anatomik b�lgelerden uygulanabilir (bkz. B�l�m 4.2).
Da��l�m:
Sa�l�kl� g�n�ll�lerde 0,25 mg/kg emicizumab�n tek bir intraven�z dozunun ard�ndan, kararl� durumda da��l�m hacmi 106 mL / kg'd�r (yani, 70 kg'l�k bir yeti�kin i�in 7,4 L).
Emicizumab�n ard���k subkutan dozlar�n� takiben hemofili A hastalar�nda pop�lasyon farmakokineti�i analizlerinden hesaplanm�� olan da��l�m hacmi (V/F) 10,4 L'dir.
Biyotransformasyon:
Emicizumab�n metabolizmas� incelenmemi�tir. IgG antikorlar� temelde lizozomal proteoliz ile katabolize edilir ve daha sonra v�cut taraf�ndan elimine edilir ya da yeniden kullan�l�r.
Eliminasyon:
Sa�l�kl� g�n�ll�lerde 0,25 mg / kg'l�k intraven�z uygulama sonras�nda, emicizumab�n toplam klerensi 3,26 mL /kg /g�n (yani, 70 kg'l�k bir yeti�kin i�in 0,228 L/d) ve ortalama terminal yar�lanma �mr� 26,7 g�n olarak belirlenmi�tir.
Sa�l�kl� g�n�ll�lerde tek subkutan enjeksiyonun ard�ndan, eliminasyon yar�lanma �mr� yakla��k 4-5 hafta olmu�tur.
Hemofili A hastalar�nda ard���k subkutan enjeksiyonlar�n ard�ndan, g�r�n�r klerens 0,272 L/g�n ve g�r�n�r eliminasyon yar�lanma �mr� 26,8 g�nd�r.
Do�rusall�k/do�rusal olmayan durum:
Emicizumab, subkutan uygulamay� takiben haftada bir kez 0,3 ila 6 mg /kg doz aral���nda hemofili A hastalar�nda doz ba��ml� bir farmakokinetik profil sergilemi�tir. Birden fazla doz maruziyeti (C) haftada 1 kez 1,5 mg/kg, 2 haftada 1 kez 3 mg/kg ve 4 haftada 1 kez 6 mg/kg i�in kar��la�t�r�labilirdir.
�zel Pop�lasyonlar:
Pediyatrik pop�lasyon
Hasta ya��n�n emicizumab�n farmakokinetik �zellikleri �zerine etkisi, 5 bebek (≥ 1 ay ila < 2 ya�), 55 �ocuk (<12 ya�) ve 50 ergeni (12 ila <18 ya�) i�eren bir hemofili A hasta pop�lasyonunun farmakokinetik analizlerinde de�erlendirilmi�tir.
Ya�, pediyatrik hastalarda emicizumab�n farmakokineti�ini etkilememi�tir. Geriyatrik pop�lasyon
Hasta ya��n�n emicizumab�n farmakokinetik �zellikleri �zerine etkisi, 65 ya� ve �zerindeki on �� g�n�ll�y� (hi�biri 77 ya��ndan b�y�k de�ildir) kapsayan bir pop�lasyonun farmakokinetik analizleri ile de�erlendirilmi�tir. G�receli biyoyararlan�m ya�la birlikte azalm��, ancak emicizumab�n farmakokinetik �zelliklerinde 65 ya� alt� ve ≥ 65 ya� bireyler aras�nda klinik olarak anlaml� farkl�l�klar g�zlenmemi�tir.
Irk
Hemofili A hastalar�nda ger�ekle�tirilen pop�lasyon farmakokinetik analizleri, �rk�n emicizumab farmakokinetik �zelliklerini etkilemedi�ini g�stermi�tir. Bu demografik fakt�r i�in doz ayarlamas� gerekli de�ildir.
B�brek yetmezli�i
B�brek yetmezli�inin emicizumab�n farmakokinetik �zellikleri �zerindeki etkileri ile ilgili �zel �al��malar y�r�t�lmemi�tir.
Pop�lasyon farmakokineti�i analizinde, hemofili A hastalar�n�n �o�unun b�brek fonksiyonunun normal oldu�u (N = 332; kreatinin klerensi [KrKl] ≥ 90 mL/dak) veya hafif b�brek yetmezli�i (N = 27; KrKl 60-89 mL/dak) oldu�u saptanm��t�r. Hafif b�brek yetmezli�i emicizumab�n farmakokineti�ini etkilememi�tir. Orta derece b�brek yetmezli�i olan hastalarda HEMLIBRA kullan�m�na ili�kin veri s�n�rl�d�r (KrKl 30-59 mL/dak olan yaln�zca 2 hasta) ve �iddetli b�brek yetmezli�i olan hastalara ili�kin veri bulunmamaktad�r. Hafif veya orta derece b�brek yetmezli�inin emicizumab�n farmakokineti�ine etkisi olup olmad��� belirlenememektedir.
Emicizumab bir monoklonal antikor olup, b�brekle at�l�mdan ziyade katabolizma ile temizlenir ve b�brek yetmezli�i olan hastalar i�in dozda de�i�iklik gerekmesi beklenmemektedir.
Karaci�er yetmezli�i
Karaci�er yetmezli�inin emicizumab�n farmakokinetik �zellikleri �zerindeki etkileri ile ilgili �zel �al��malar y�r�t�lmemi�tir. Pop�lasyon farmakokinetik analizlerinde yer alan hemofili A hastalar�n�n �o�unda karaci�er fonksiyonlar� normaldir (bilirubin ve AST ≤ Normal �st S�n�r (N�S), n = 300) veya hafif karaci�er yetmezli�i mevcuttur (bilirubin ≤ N�S ve AST> N�S veya bilirubin <1 ila 1,5 × N�S ve herhangi bir d�zeyde AST, n=51). Sadece 6 hastada orta derece karaci�er yetmezli�i (1,5 x N�S < bilirubin ≤ 3 x N�S ve herhangi bir d�zeyde AST) saptanm��t�r. Hafif karaci�er yetmezli�i, emicizumab�n farmakokinetik �zelliklerini etkilememi�tir (bkz. B�l�m 4.2). Emicizumab�n g�venlili�i ve etkilili�i karaci�er yetmezli�i olan hastalarda �zel olarak test edilmemi�tir. Hafif ila orta �iddette karaci�er yetmezli�i olan hastalar klinik �al��malara dahil edilmi�tir. �iddetli karaci�er yetmezli�i olan hastalarda HEMLIBRA kullan�m�na ili�kin veri yoktur.
Emicizumab bir monoklonal antikor olup, karaci�er metabolizmas�ndan ziyade katabolizma ile temizlenir ve karaci�er yetmezli�i olan hastalar i�in dozda de�i�iklik gerekmesi beklenmemektedir.
Di�er �zel pop�lasyonlar
Modelleme �al��mas�, hipoalbuminemi ve ya�lar�na g�re d���k v�cut a��rl��� olan hastalara daha nadir verilen dozlar�n daha d���k emicizumab maruziyetini g�stermektedir; sim�lasyonlar da bu hastalarda klinik olarak anlaml� kanama kontrol� sa�lanabilece�ine i�aret etmektedir.
5.3. Klinik �ncesi g�venlilik verileri
5.3. Klinik �ncesi g�venlilik verileri
Fertilite
Emicizumab, 30 mg/kg/haftal�k en y�ksek test edilen doza (EAA temelinde 3 mg/kg/haftal�k en y�ksek dozda insan maruziyetinin 11 kat�na e�de�er) kadar erkek veya di�i sinomolgus maymunlar�n�n �reme organlar�nda herhangi bir de�i�ikli�e neden olmam��t�r.
Teratojenisite
Emicizumab�n embriyo-f�tal geli�im �zerindeki potansiyel yan etkileri ile ilgili veri mevcut de�ildir.
Enjeksiyon yeri reaksiyonlar�
Subkutan enjeksiyon sonras�nda hayvanlarda tersine �evrilebilir hemoraji, perivask�ler monon�kleer h�cre infiltrasyonu, subkutis dejenerasyonu/nekrozu ve subkutiste endotelyum �i�mesi bildirilmi�tir.
6. FARMAS�T�K �ZELL�KLER
6.1. Yard�mc� maddelerin listesi
L-Arjinin L-Histidin
L-Aspartik asit Poloksamer 188 Enjeksiyonluk su
6.2. Ge�imsizlikler
HEMLIBRA ile �nerilen enjekt�rler ve i�neler aras�nda ge�imsizlik g�zlenmemi�tir (bkz. B�l�m 6.6).
Ge�imlilik �al��malar� olmad���ndan, bu t�bbi �r�n di�er t�bbi �r�nlerle kar��t�r�lmamal�d�r.
6.3. Raf �mr�
A��lmam�� flakon 24 ay.Buzdolab�ndan ��kar�ld���nda, a��lmam�� flakonlar 7 g�ne kadar oda s�cakl���nda (30°C'nin alt�nda) saklanabilir.
Oda s�cakl���nda sakland�ktan sonra, a��lmam�� flakonlar buzdolab�na geri konulabilir. Oda s�cakl���nda k�m�latif saklama s�resi 7 g�n� ge�memelidir. Flakonlar asla 30°C'yi a�an s�cakl�klara maruz b�rak�lmamal�d�r. Oda s�cakl���nda 7 g�nden uzun s�re bekleyen veya 30°C'yi a�an s�cakl�klara maruz kalan flakonlar imha edilmelidir.
Delinmi� flakon ve doldurulmu� enjekt�r
Mikrobiyolojik a��dan, flakondan enjekt�re transfer edildikten sonra t�bbi �r�n hemen kullan�lmal�d�r. Hemen kullan�lmazsa, kullanacak ki�i kullan�m s�ras�ndaki saklama zaman�ndan ve ko�ullar�ndan sorumludur.
6.4. Saklamaya y�nelik �zel tedbirler
Flakonlar� 2°C-8°C'de buzdolab�nda saklay�n�z. Dondurmay�n�z. �alkalamay�n�z. Flakonu, ���ktan korumak i�in kutusunda saklay�n�z.
T�bbi �r�n ilk defa a��ld�ktan sonraki saklama ko�ullar� i�in bkz. B�l�m 6.3.
6.5. Ambalaj�n niteli�i ve i�eri�i
1 mL HEMLIBRA ��zeltisi (30 mg/mL) i�eren, bir floro-re�ine film ile lamine edilmi� ve plastik bir ge�me disk oturtulmu� bir al�minyum ba�l�kla b�k�lerek kapat�lm�� b�til lastik t�pal� bir adet 3 mL �effaf cam tip I flakon. Her kartonda 1 flakon bulunmaktad�r.
6.6. Be�eri t�bbi �r�nden arta kalan maddelerin imhas� ve di�er �zel �nlemler
HEMLIBRA ��zeltisi, seyreltilmesine gerek olmayan, subkutan enjeksiyon i�in steril, koruyucu i�ermeyen ve kullan�ma haz�r bir ��zeltidir.
HEMLIBRA, uygulama �ncesinde herhangi bir partik�ll� madde veya renk de�i�ikli�inin olmad���ndan emin olmak i�in g�rsel olarak incelenmelidir. HEMLIBRA, renksiz ile hafif sar� aras� renkte bir ��zeltidir. Partik�ll� maddeler g�r�lebiliyorsa veya �r�n�n rengi de�i�irse HEMLIBRA ��zeltisi at�lmal�d�r.
HEMLIBRA �alkalanmamal�d�r.
HEMLIBRA enjeksiyonluk ��zelti �i�eleri yaln�zca tek kullan�ml�kt�r.
HEMLIBRA ��zeltisini flakondan �ekip subkutan yoldan enjekte etmek i�in bir enjekt�r, transfer i�nesi ve bir enjeksiyon i�nesi gereklidir.
1 mL'ye kadar HEMLIBRA ��zeltisinin enjekte edilmesi i�in 1 mL'lik bir enjekt�r kullan�lmal�d�r; buna kar��l�k 1 mL'den fazla ve en fazla 2 mL'ye kadar olan bir enjeksiyon i�in 2 - 3 mL'lik bir enjekt�r kullan�lmal�d�r.
Farkl� dozlarda flakonlar�n ayn� enjekt�rde kullan�m� i�in HEMLIBRA “Kullanma Talimat�”na bak�n�z. Re�etelenmi� dozu almak i�in farkl� dozlarda flakonlar birlikte kullan�ld���nda, farkl� HEMLIBRA konsantrasyonlar� (30 mg/mL ve 150 mg/mL) kullan�lmamal�d�r.
Uygulama ile ilgili ilave bilgiler i�in l�tfen B�l�m 4.2 ve kullanma talimat�na bak�n�z (Kullanma Talimat�'n�n sonunda yer alan ‘Uygulama talimatlar�'na bak�n�z).
Kullan�lmam�� olan �r�nler ya da at�k materyaller, “T�bbi At�klar�n Kontrol� Y�netmeli�i'' ve “Ambalaj At�klar�n�n Kontrol� Y�netmeli�i”ne uygun olarak imha edilmelidir.
 Artrit
Artrit, olduk�a yayg�n bir hastal�kt�r ancak iyi anla��lamam��t�r. Asl�nda �artrit� tek bir hastal���n ad� de�ildir; eklem a�r�s� veya eklem hastal�klar�n� adland�rman�n gayri resmi yoludur.
Artrit
Artrit, olduk�a yayg�n bir hastal�kt�r ancak iyi anla��lamam��t�r. Asl�nda �artrit� tek bir hastal���n ad� de�ildir; eklem a�r�s� veya eklem hastal�klar�n� adland�rman�n gayri resmi yoludur. |
 Ruh ve Ak�l Sa�l���m�z� Geli�tirmek
�yi ak�l ve ruh sa�l��� sahip olmaktan ziyade, yapt���n�z �eylerdir. Ak�l ve
ruhsal olarak sa�l�kl� olmak i�in kendinize de�er vermeli ve kendinizi kabul
etmelisiniz.
Ruh ve Ak�l Sa�l���m�z� Geli�tirmek
�yi ak�l ve ruh sa�l��� sahip olmaktan ziyade, yapt���n�z �eylerdir. Ak�l ve
ruhsal olarak sa�l�kl� olmak i�in kendinize de�er vermeli ve kendinizi kabul
etmelisiniz. |
 |
Travma Sonras� Bunal�m� Travmatik bir olay, g�nl�k ola�an olaylar�n d���nda olan ve ki�iyi derinden rahats�z eden bir olayd�r.Bir�ok olay b�yle bir etki g�sterebilir. |
 |
A��r� Alkol Kullan�m�, Alkolizm Alkol ba��ml�l���, alkol kullan�m� ve alkol sorunlar� aras�ndaki fark� a��klamak g��t�r. �rne�in, ge�mi�te alkol kullanm�� olan bir kimsenin mutlaka alkol ba��ml�s� olmas� gerekmez. |
 |
Omurilik zedelenmeleri Omurilik zedelenmesini takip eden birka� g�n i�inde, hi�kimse hasarin ne kadar olacagini tahmin edemez. Buradaki sorun, omuriligin herhangi bir zedelenmesinden hemen sonra, bir omurilik sokunun olusmasidir. |
�LA� GENEL B�LG�LER�
Roche M�stahzarlar� Sanayi A.�.
| Sat�� Fiyat� | 79537.95 TL [ 3 Aug 2026 ] |
| �nceki Sat�� Fiyat� | 79537.95 TL [ 27 Jul 2026 ] |
| Original / Jenerik | Original �la� |
| Re�ete Durumu | K�rm�z� Re�eteli bir ila�d�r. |
| Barkodu | 8699505773544 |
| Etkin Madde | Emisizumab |
| ATC Kodu | B02BX06 |
| Birim Miktar | 30 |
| Birim Cinsi | MG/ML |
| Ambalaj Miktar� | 1 |
| Kan ve Kan Yap�c� Organlar > K Vitamini ve Di�er Hemostatikler |
| �thal ( ref. �lke : Isvicre ) ve Be�eri bir ila�d�r. |