ADCETRIS 50 mg IV inf�zyonluk ��zelti konsantresi i�eren flakon (1 flakon) K�sa �r�n Bilgisi
{ Brentuksimab Vedotin }
1. BE�ER� TIBB� �R�N�N ADI
ADCETR�S 50 mg IV inf�zyonluk ��zelti konsantresi i�in toz i�eren flakon Steril, Sitotoksik
2. KAL�TAT�F VE KANT�TAT�F B�LE��M
Etkin madde
Her bir flakon 50 mg brentuximab vedotin i�erir.
Kullan�ma haz�rland�ktan sonra (bkz. b�l�m 6.6), her mL'si 5 mg brentuximab vedotin i�erir.
ADCETR�S, antimikrot�b�l ajan monometil auristatin E'ye (MMAE) kovalent ba�l� bir CD30 g�d�ml� monoklonal antikordan (�in hamster� over h�crelerinde rekombinant DNA teknolojisi ile �retilen rekombinant kimerik imm�noglobulin G1 [IgG1]) olu�an bir antikor-ila� konj�gat�d�r.
Yard�mc� maddeler
Sodyum sitrat dihidrat 56,1 mg
Yard�mc� maddelerin tam listesi i�in bkz. b�l�m 6.1.
3. FARMAS�T�K FORMU
�nf�zyonluk ��zelti konsantresi tozu. Beyaz ila k�r�k beyaz kek veya toz.
4. KL�N�K �ZELL�KLER
4.1. Terap�tik endikasyonlar
Hodgkin lenfoma (HL)
ADCETR�S, Evre IV CD30 pozitif klasik Hodgkin lenfoma tan�s� konmu� olan hastalardan;
60 ya� �zerinde olup, n�ropatisi bulunmayan IPS≥4 olgularda,
4.2. Pozoloji ve uygulama �ekli
ADCETR�S, anti-kanser ajanlar�n�n kullan�m�nda deneyimli bir hekimin g�zetimi alt�nda uygulanmal�d�r.
Pozoloji / uygulama s�kl��� ve s�resi
�nceden Tedavi Edilmemi� Hodgkin lenfoma
Kemoterapi ile kombinasyon halinde (doksorubisin [A], vinblastin [V] ve dakarbazin [D] [AVD]) �nerilen doz, her bir siklusun 28 g�n oldu�u toplam 6 siklus s�ren bir tedavide her bir siklusun 1. ve 15. g�nlerinde 30 dakika s�reyle intraven�z inf�zyon �eklinde uygulanan 1.2 mg/kg'd�r (bkz. b�l�m5.1).
Kombinasyon tedavisi alan, daha �nce tedavi edilmemi� Hodgkin lenfomal� t�m hastalar�n tedavisi i�in ilk dozdan ba�layarak b�y�me fakt�r� (G-CSF) ile desteklenen primer profilaksi �nerilmektedir.(bkz. b�l�m
4.4.).
Hodgkin lenfomal� (HL) hastalar�n birinci basamak tedavisinde ADCETR�S ile birlikte verilen kemoterapi ajanlar�n�n k�sa �r�n bilgilerine (K�B) bak�n�z.
N�ks veya progresyon riski artan Hodgkin lenfoma (HL)
�nerilen doz, 3 haftada bir 30 dakika s�reyle intraven�z inf�zyon �eklinde uygulanan 1,8 mg/kg'd�r. ADCETR�S tedavisine, klinik yarg�ya g�re, otolog k�k h�cre nakline ba�l� iyile�me s�recinden sonra ba�lanmal�d�r. Bu hastalar 16 siklusa kadar tedavi almal�d�r (bkz. b�l�m 5.1.)
N�kseden veya tedaviye diren�li Hodgkin lenfoma (HL)
�nerilen doz, 3 haftada bir 30 dakika s�reyle intraven�z inf�zyon �eklinde uygulan an 1,8 mg/kg'd�r.
Daha �nce ADCETR�S tedavisine yan�t veren hastalar i�in tavsiye edilen ba�lang�� dozu her 3 haftada bir 30 dakika boyunca intraven�z inf�zyon olarak uygulanan 1,8 mg/kg'd�r. Alternatif olarak, tedavi tolere edilmi� en son doz ile ba�lat�labilir (bkz. b�l�m 5.1).
Hastal�k progrese olana ya da kabul edilemez d�zeyde toksisite meydana gelene kadar tedaviye devam edilmelidir (bkz. b�l�m 4.4).
Stabil hastal��a ya da daha iyid�zeye ula�an hastalar en az 8 siklus ve en fazla 16 siklus (yakla��k 1 y�l) tedavi
almal�d�r (bkz. b�l�m5.1).
N�kseden veya tedaviye diren�li sistemik anaplastik b�y�k h�creli lenfoma (sALCL)
�nerilen doz, 3 haftada bir 30 dakika s�reyle intraven�z inf�zyon �eklinde uygulan an 1,8 mg/kg'd�r.
Daha �nce ADCETR�S tedavisine yan�t veren hastalar i�in tavsiye edilen ba�lang�� dozu her 3 haftada bir 30 dakika boyunca intraven�z inf�zyon olarak uygulanan 1,8 mg/kg'd�r. Alternatif olarak, tedavi tolere edilmi� en son doz ile ba�lat�labilir (bkz. b�l�m 5.1).
Hastal�k progrese olana ya da kabul edilemez d�zeyde toksisite meydana gelene kadar tedaviye devam edilmelidir (bkz. b�l�m 4.4).
Stabil hastal��a ya da daha iyid�zeye ula�an hastalar en az 8 siklus ve en fazla 16 siklus (yakla��k 1 y�l) tedavi almal�d�r (bkz. b�l�m5.1).
Kutan�z T-h�creli lenfoma (CTCL)
�nerilen doz, 3 haftada bir 30 dakika s�reyle intraven�z inf�zyon �eklinde uygulanan 1,8 mg/kg'd�r. CTCL'si olan hastalara 16 siklusa kadar uygulanmal�d�r (bkz. b�l�m 5.1).
�nceden tedavi edilmemi� sistemik anaplastik b�y�k h�creli lenfoma (sALCL) ve di�er CD30 pozitifli�i olan periferik T-h�creli lenfomalar (PTCL)
Kemoterapi ile kombinasyon tedavisinde (siklofosfamid [C], doksorubisin [H] ve prednizon [P] [CHP]) tavsiye edilen doz, 3 haftada bir 6 ila 8 siklusolarak, en fazla 180 mg olacak �ekilde, 30 dakika s�reyle intraven�z inf�zyon olarak uygulanan 1.8 mg/kg'd�r (Bkz. b�l�m 5.1)
Kombinasyon tedavisi alan, daha �nce tedavi edilmemi� t�m sALCL ve di�er CD30 pozitiflik belirlenen
PTCL hastalar�n�n tedavisi i�in ilk dozdan itibaren b�y�me fakt�r� (G-CSF) ile desteklenen primer profilaksi �nerilmektedir (bkz. b�l�m 4.4.).
Daha �nce tedavi edilmemi� sALCL ve di�er CD30 pozitiflik belirlenen PTCL hastalar� i�in ADCETR�S ile birlikte uygulanan kemoterapi ajanlar�n�n k�sa �r�n bilgilerine bak�n�z.
Genel
E�er hastan�n beden a��rl��� 100 kg'�n �zerinde ise doz hesaplamas�nda 100 kg kullan�lmal�d�r (bkz. b�l�m 6.6).
Bu tedavinin her bir dozunun uygulanmas�ndan �nce tam kan say�m� izlemi yap�lmal�d�r (bkz. b�l�m 4.4).
Hastalar, inf�zyon s�ras�nda ve sonras�nda izlenmelidir (bkz. b�l�m 4.4). Doz ayarlamalar�
N�tropeni
E�er tedavi s�ras�nda n�tropeni geli�irse, bu durum dozlar ertelenerek giderilmelidir. S�ras�yla monoterapi ve kombinasyon terapisinde uygun doz uygulamas� �nerileri i�in a�a��da, Tablo 1 ve Tablo 2'ye bak�n�z (ayr�ca bkz. b�l�m 4.4).
Tablo 1: Monoterapi olarak n�tropeni i�in doz uygulamas� �nerileri
N�tropeninin �iddet derecesi (belirti ve semptomlar [k�salt�lm�� CTCAEtan�m�]) | Doz uygulamas� program�nda d�zenleme |
Derece 1 (<LLN - 1500/mm3 <LLN - 1,5 x 109/L) veya | Ayn� doza ve doz program�na devam edilir |
Derece 2 (<1500 - 1000/mm3 <1,5 – 1,0 x 109/L) | |
Derece 3 (<1,000 - 500/mm3 <1,0 - 0,5 x 109/L) veya | Toksisite ≤ Derece 2 olana veya ba�lang��taki d�zeye d�nene kadar |
Derece 4 (<500/mm3 <0,5 x 109/L) | dozlara ara verilir, ard�ndan ayn� doz ve program ile tedaviye kald��� yerden devam edilir . |
| Derece 3 veya Derece 4 n�tropeni geli�en hastalar i�in sonraki sikluslarda G-CSF veya GM-CSF verilmesi d���n�l�r. |
Tablo 2: Kombinasyon tedavisi s�ras�nda n�tropeni i�in doz �nerileri
N�tropeni �iddeti derecesi (belirti ve semptomlar [k�salt�lm�� CTCAEtan�m�]) | Doz uygulama program�n�n de�i�tirilmesi |
Derece 1 (< LLN-1500/mm < LLN-1.5 x 10/L) veya Derece 2 (< 1500-1000/mm < 1.5-1.0 x 10/L) Derece 3 (< 1,000-500/mm < 1.0-0,5 x 10/L) veya Derece 4 (< 500/mm < 0,5 x 10/L) | Kombinasyon tedavisi alan t�m yeti�kin hastalar i�in ilk dozdan ba�layarak G-CSF ile primer profilaksi �nerilir. Ayn� doza ve doz program�na devam edilir. |
Periferik n�ropati
E�er tedavi s�ras�nda periferik duyusal veya motor n�ropatiortaya ��kar yada k�t�le�irse, s�ras�yla monoterapi ve kombinasyon tedavisi i�in uygun doz uygulamas� �nerileri i�in a�a��da, Tablo 3 ve Tablo 4'e bak�n�z (bkz. b�l�m 4.4).
Tablo 3: Monoterapide yeni veya k�t�le�en periferik duyusal veya motor n�ropati i�in doz uygulamas� �nerileri
Periferik duyusal veya motor n�ropati �iddeti (i�aretler ve semptomlar [k�salt�lm�� CTCAEa tan�m�]) | Doz ve programda d�zenleme |
Derece 1 (parestezi ve/veya refleks kayb� var, i�lev kayb� yok) | Ayn� doza ve programa devam edilir. |
Derece 2 (i�levleri engelliyor fakat g�nl�k ya�am aktivitelerini etkilemiyor) | Toksisite ≤ Derece 1 olana veya ba�lang��taki d�zeye d�nene kadar dozlara ara verilir, ard�ndan tedaviye 3 haftada bir 1,2 mg/kg (maksimum 120 mg) �eklinde azalt�lm�� doz ile devam edilir |
Derece 3 (g�nl�k ya�am aktivitelerini etkiliyor) | Toksisite ≤ Derece 1 olana veya ba�lang��taki d�zeye d�nene kadar dozlara ara verilir, ard�ndan tedaviye 3 haftada bir en fazla 120 mg olacak �ekilde 1,2 mg/kg olarak azalt�lm�� doz ile devam edilir. |
Derece 4 (engelleyici duyusal n�ropati veya hayat� tehdit edici ya da felce giden motor n�ropati) | Tedavi kesilir |
Tablo 4: Kombinasyon tedavisi s�ras�nda yeni veya k�t�le�en periferik duyusal veya motor n�ropati i�in doz �nerileri
| AVD ile kombinasyon Tedavisi | CHP ile kombinasyon tedavisi |
Periferik duyusal veya motor n�ropatinin �iddeti (belirti ve semptomlar [k�salt�lm�� CTCAEtan�m�]) | Doz ve programda d�zenleme | Doz ve programda d�zenleme |
Derece 1 (parestezi ve / veya refleks kayb� var, i�lev kayb� yok) | Ayn� doz ve programla devam edilir. | Ayn� doz ve programla devam edilir. |
Derece 2 (i�levleri engelliyor fakat | Doz 2 haftada bir | Duyusal n�ropati: Ayn� |
g�nl�k ya�am aktivitelerini etkilemiyor) | maksimum 90 mg olacak �ekilde 0,9 mg/kg'a kadar d���r�l�r. | doz seviyesinde tedaviye devam edilir. Motor n�ropati: Doz her 3 haftada bir maksimum 120 mg olacak �ekilde |
|
| 1,2 mg/kg'a d���r�l�r. |
Derece 3 (g�nl�k ya�am | Toksisite ≤ Derece 2 | Duyusal n�ropati: Doz |
aktivitelerini etkiliyor) | olana kadar ADCETR�S ile tedaviye ara verilir, | her 3 haftada bir maksimum 120 mg |
| daha sonra her 2 haftada bir maksimum 90 mg | olacak �ekilde 1,2 mg/kg'a d���r�l�r. |
| olacak �ekilde 0,9 mg/kg'a d���r�lm�� bir dozda tedaviye yeniden | Motor n�ropati: Tedavi kesilir. |
| ba�lan�r. |
Derece 4 (engelleyici duyusal n�ropati veya hayat� tehdit edici ya da felce giden motor n�ropati) | Tedavi kesilir. | Tedavi kesilir. |
Uygulama �ekli
�nerilen ADCETR�S dozu, 30 dakika s�reyle inf�zyon yoluyla uygulan�r. Uygulama �ncesinde bu t�bbi �r�n�n kullan�ma haz�rlanmas� ve seyreltilmesi ile ilgili talimatlar i�in bkz. b�l�m 6.6.
ADCETR�S h�zl� intraven�z inf�zyon veya bolus �eklinde uygulanmamal�d�r. ADCETR�S bu amaca tahsis edilmi� bir intraven�z hat yoluyla uygulanmal�d�r ve di�er t�bbi �r�nler ile kar��t�r�lmamal�d�r (bkz. b�l�m 6.2).
�zel pop�lasyonlara ili�kin ek bilgiler:
B�brek/Karaci�er yetmezli�i:
Kombinasyon tedavisi
B�brek yetmezli�i olan hastalar advers olaylar a��s�ndan yak�ndan izlenmelidir. Serum kreatinin seviyesinin ≥2,0 mg/dL veya kreatinin klerensi veya hesaplanan kreatinin klerensinin ≤40 mL/dakika oldu�u b�brek yetmezli�i olan hastalarda ADCETR�S ile kombinasyon kemoterapisine y�nelik klinik �al��ma deneyimi mevcut de�ildir. �iddetli b�brek yetmezli�i olan hastalarda ADCETR�S'in kemoterapi ile kombine kullan�m�ndan ka��n�lmal�d�r.
Karaci�er yetmezli�i olan hastalar advers olaylar a��s�ndan yak�ndan izlenmelidir. Hafif karaci�er yetmezli�i olan ve ADCETR�S'i AVD ile birlikte kullanan hastalarda �nerilen ba�lang�� dozu, 2 haftada bir 30 dakikas�reyle intraven�z inf�zyonla verilen 0,9 mg/kg'd�r. Hafif karaci�er yetmezli�i olan ve ADCETR�S'i CHP ile birlikte kullanan hastalarda �nerilen ba�lang�� dozu, 3 haftada bir 30 dakika s�reyle intraven�z inf�zyonla verilen 1,2 mg/kg'd�r. Toplam biluribinin normal �st limit de�erinin (ULN) 1,5 kat�ndan fazla oldu�u (e�er Gilbert Sendromuna ba�l� de�ilse) veya, aspartat aminotransferaz (AST) veya alanin aminotransferaz (ALT) de�erleri normal �st limit de�erinin (ULN) 3 kat�ndan fazla oldu�u veya karaci�erdeki y�kselen de�erlerin HL varl���na ba�l� olabilece�i durumlarda normal �st limitde�erinin (ULN) 5 kat�ndan fazla oldu�u �iddetli karaci�er yetmezli�i olan hastalarda ADCETR�S ile kombinasyon kemoterapisine y�nelik klinik �al��ma deneyimi mevcut de�ildir. Orta veya �iddetli karaci�er yetmezli�i olan hastalarda ADCETR�S kemoterapi ile kombinasyon tedavisi �eklinde kullan�lmamal�d�r.
Monoterapi
�iddetli b�brek yetmezli�i olan hastalarda �nerilen ba�lang�� dozu, 3 haftada bir 30 dakika s�reyle intraven�z inf�zyon �eklinde uygulanan 1,2 mg/kg'd�r. B�brek yetmezli�i olan hastalar advers olaylar a��s�ndan yak�ndan izlenmelidir (bkz. b�l�m5.2).
Karaci�er yetmezli�i olan hastalarda �nerilen ba�lang�� dozu, 3 haftada bir 30 dakika s�reyle intraven�z inf�zyon �eklinde uygulanan 1,2 mg/kg'd�r. Karaci�er yetmezli�i olan hastalar advers olaylar a��s�ndan yak�ndan izlenmelidir (bkz. b�l�m5.2).
Pediyatrik pop�lasyon:
18 ya��n alt�ndaki pediyatrik hastalarda ADCETR�S'in g�venlilik ve etkilili�i hen�z ispatlanmam��t�r. Mevcut g�ncel veriler b�l�m 4.8, 5.1 ve 5.2'de sunulmu�tur ancak pozolojiye y�nelik bir tavsiye verilememektedir.
Geriyatrik pop�lasyon:
65 ya� ve �zeri hastalar i�in �nerilen doz yeti�kinler ile ayn�d�r. Mevcut g�ncel veriler b�l�m 4.8,
5.1 ve 5.2'de sunulmu�tur.
4.3. Kontrendikasyonlar
Etkin madde
4.4. �zel kullan�m uyar�lar� ve �nlemleri
Progresif multifokal l�koensefalopati
ADCETR�S ile tedavi edilen hastalarda John Cunningham Vir�s� (JCV) reaktivasyonu sonucu progresif multifokal l�koensefalopati (PML) ve �l�m meydana gelebilir. �nceden �oklu kemoterapi rejimleri ald�ktan sonra bu tedaviyi g�ren hastalarda PML bildirilmi�tir. PML latent, JCV reaktivasyonu sonucu ortaya ��kan ve s�kl�kla �l�mle sonu�lanan, nadir, demiyelinizan bir santral sinir sistemi hastal���d�r.
Hastalar, PML'ye i�aret edebilecek yeni veya k�t�le�en n�rolojik, bili�sel veya davran��sal belirtiler ve semptomlar a��s�ndan yak�ndan izlenmelidir. T�m ��pheli PML olgular�nda ADCETR�S doz uygulamalar� durdurulmal�d�r. PML de�erlendirmesi i�in �neriler n�roloji kons�ltasyonunu, beyine y�nelik gadolinyum bazl� kontrast manyetik rezonans g�r�nt�leme incelemesini ve polimeraz zincir reaksiyonu yoluyla beyin-omurilik s�v�s�nda JCV DNA analizini ya da JCV kan�t� i�in beyin biyopsisini i�ermektedir. JCV PCR'nin negatif olmas� PML olas�l���n� ekarte etmez. �ayet alternatif tan� koyulam�yorsa ek izlem ve de�erlendirme gerekebilir. PML tan�s� do�ruland��� takdirde ADCETR�S dozlar� kesilmeli ve bir daha ba�lanmamal�d�r.
Hekim, �zellikle, PML'ye i�aret eden ve hastan�n fark edemeyebilece�i semptomlar konusunda dikkatli olmal�d�r (�rn., bili�sel, n�rolojik veya psikiyatrik semptomlar).
Pankreatit
ADCETR�S ile tedavi edilen hastalarda akut pankreatit g�zlenmi�tir. �l�mle sonu�lanan vakalar
bildirilmi�tir.
Hastalar, akut pankreatite i�aret edebilecek yeni veya k�t�le�en abdominal a�r� a��s�ndan yak�ndan izlenmelidir. Hastan�n de�erlendirmesi fizik muayene, serum amilaz ve serum lipaza y�nelik laboratuvar incelemesi ve ultrason gibi abdominal g�r�nt�leme y�ntemlerini ve di�er uygun tan�sal y�ntemleri i�erebilir. T�m��pheli akut pankreatit olgular�nda ADCETR�S'e ara verilmelidir. Akut pankreatit tan�s� do�ruland��� takdirde ADCETR�S kesilmelidir.
Pulmoner toksisite
ADCETR�S alan hastalarda, baz�lar� �l�mle sonu�lanan pn�monit, interstisyel akci�er hastal��� ve akut solunum distres sendromunu(ARDS) i�eren pulmoner toksisite vakalar� bildirilmi�tir.
Brentuximab ile bir nedensellik ili�kisi kurulamam�� ise de pulmoner toksisite riski g�z ard� edilemez. Yeni pulmoner semptomlar�n ortaya ��kmas� veya var olan semptomlar�n k�t�le�mesi durumunda (�rn: �ks�r�k, dispne) derhal bir tan� de�erlendirmesi ger�ekle�tirilmeli ve hastalar uygun �ekilde tedavi edilmelidir. De�erlendirme s�ras�nda ve semptomlar iyile�ene kadar ADCETR�S uygulanmas�na ara verilmesi de�erlendirilmelidir.
Ciddi enfeksiyonlar ve f�rsat�� enfeksiyonlar
ADCETR�S ile tedavi edilen hastalarda pn�moni, stafilokokal bakteriyemi, sepsis/septik �ok (�l�mle sonu�lanan vakalar dahil) ve herpes zoster, sitomegalovirus (CMV) (reaktivasyon) gibi ciddi enfeksiyonlar ile Pneumocystis jiroveci pn�monisi ve oral kandidiyaz gibi f�rsat�� enfeksiyonlar bildirilmi�tir. Hastalar tedavi s�resince olas� ciddi ve f�rsat�� enfeksiyonlar�n ortaya ��kmas� a��s�ndan dikkatle izlenmelidir.
�nf�zyona ba�l� reaksiyonlar
Ani ve gecikmi� inf�zyona ba�l� reaksiyonlar (�BR) ve ayr�ca anafilaktik reaksiyonlar bildirilmi�tir.
Hastalar inf�zyon s�ras�nda ve sonras�nda dikkatle izlenmelidir. Anafilaktik reaksiyon olu�mas� durumunda ADCETR�S uygulamas� derhal ve bir daha ba�lanmamak �zere kesilmeli ve uygun t�bbi tedavi ba�lat�lmal�d�r.
E�er inf�zyona ba�l� reaksiyon ortaya ��karsa, inf�zyona ara verilmeli ve uygun t�bbi tedavi ba�lat�lmal�d�r. Semptom giderildikten sonra inf�zyona daha d���k bir h�zda tekrar ba�lanabilir. �nceden inf�zyona ba�l� bir reaksiyon ya�am�� olan hastalara, m�teakip inf�zyonlar i�in �n ila� tedavisi uygulanmal�d�r. �n ila� tedavisi parasetamol, bir antihistaminik ve bir kortikosteroid i�erebilir.
ADCETR�S'e kar�� antikorlar� olan hastalarda inf�zyona ba�l� reaksiyonlar daha s�k ve daha �iddetlidir (bkz. b�l�m 4.8).
T�m�r lizis sendromu
ADCETR�S ile ili�kili t�m�r lizis sendromu (TLS) bildirilmi�tir. H�zl� b�y�yen t�m�r� ve y�ksek t�m�r y�k� olan hastalar, t�m�r lizis sendromu a��s�ndan daha y�ksek risk alt�ndad�r. Bu hastalar yak�ndan izlenmeli ve en iyi t�bbi uygulamalar do�rultusunda tedavi uygulanmal�d�r. TLS tedavisi agresif hidratasyon, renal fonksiyon izlemi, elektrolit anormalliklerinin d�zeltilmesini, anti- hiper�risemik tedaviyi ve destekleyici bak�m� i�erebilir.
Periferik n�ropati
ADCETR�S tedavisi hem duyusal hem de motorperiferik n�ropatiye neden olabilir. ADCETR�S ile ind�klenen periferik n�ropati tipik olarak bu t�bbi �r�n�n k�m�latif maruziyetinin bir etkisi olup, �o�u durumda geri d�n���ml�d�r.
Klinik �al��malarda, hastalar�n �o�unda, hastal���n semptomlar�nda gerileme veya iyile�me g�zlenmi�tir (bkz. b�l�m 4.8). Hastalar hipoestezi, hiperestezi, parestezi, rahats�zl�k, yanma hissi, n�ropatik a�r� veya g��s�zl�k gibi n�ropati semptomlar� a��s�ndan izlenmelidir. Yeni veya k�t�le�en periferik n�ropati ya�ayan hastalarda ADCETR�S dozunda erteleme ve azaltma veya tedavinin b�rak�lmas� gerekebilir (bkz. b�l�m4.2).
Hematolojik toksisiteler
ADCETR�S ile Derece 3 veya Derece 4 anemi, trombositopeni ve uzun s�reli (≥1 hafta) Derece 3 veya Derece 4 n�tropeni g�r�lebilir. Her dozun uygulanmas�ndan �nce tam kan say�m� izlemi yap�lmal�d�r. Derece 3 veya Derece 4 n�tropeni geli�mesi durumunda, bkz. b�l�m 4.2. Febril n�tropeni ADCETR�S ile tedavi sonucu febril n�tropeni bildirilmi�tir (klinik veya mikrobiyolojik olarak belgelenmi� enfeksiyon yoklu�unda , n�trofil say�s� <1,0 x 109/L, ate� ≥38,5°C ile birlikte k�keni bilinmeyen ate�; ref. CTCAE v3). Bu tedavinin her bir dozunun uygulanmas�ndan �nce tam kan say�m� izlemi yap�lmal�d�r. Hastalar, ate� a��s�ndan yak�ndan izlenmelidir ve febril n�tropeni geli�ti�i takdirde en iyi t�bbi uygulamalar do�rultusunda tedavi uygulanmal�d�r.
AVD veya CHP ile kombinasyon tedavisinde ileri ya� febril n�tropeni i�in bir risk fakt�r�d�r. ADCETR�S, AVD veya CHP ile kombine olarak verildi�inde ilk dozdan itibaren ba�layacak �ekilde ya�a bak�lmaks�z�n t�m yeti�kin hastalara G-CSF ile primer profilaksisi �nerilmektedir.
�iddetli kutan�z advers reaksiyonlar (SCAR'lar)
ADCETR�S ile Stevens-Johnson sendromu (SJS), toksik epidermal nekroliz (TEN) ve eozinofili ve sistemik semptomlu ila� reaksiyonlar� (DRESS) dahil olmak �zere �iddetli kutan�z advers reaksiyonlar (SCAR'lar) bildirilmi�tir. SJS ve TEN i�in �l�mle sonu�lanan vakalar bildirilmi�tir. SJS, TEN veya DRESS ortaya ��karsa, ADCETR�S tedavisi kesilmeli ve uygun t�bbi tedavi uygulanmal�d�r.
Gastrointestinal komplikasyonlar
ADCETR�S ile tedavi edilen hastalarda, baz�lar� �l�mle sonu�lanan ba��rsak t�kanmas�, ileus, enterokolit, n�tropenik kolit, erozyon, �lser, perforasyon ve hemoraji i�eren gastrointestinal komplikasyonlar rapor edilmi�tir. Yeni gastrointestinal semptomlar�n ortaya ��kmas� veya var olanlar�n k�t�le�mesi durumunda derhal tan� de�erlendirmesi yap�l�p uygun tedavi ger�ekle�tirilmelidir.
Hepatotoksisite
ADCETR�S ile alanin aminotransferaz (ALT) ve aspartat aminotransferaz (AST) y�kselmesi �eklinde hepatotoksisite rapor edilmi�tir. Baz�lar� �l�mle sonu�lanan ciddi hepatotoksisite vakalar� da meydana gelmi�tir. �nceden var olan karaci�er hastal���, komorbiditeler ve e�zamanl� olarak kullan�lan ila�lar da bu riski art�rabilir. Tedavi ba�lat�lmadan �nce karaci�er fonksiyon testi yap�lmal� ve ADCETR�S alan hastalarda rutin olarak izlenmelidir. Hepatotoksisite ya�ayan hastalarda dozun geciktirilmesi veya de�i�tirilmesi ya da ADCETR�S'in kesilmesi gerekebilir.
Hiperglisemi
Klinik �al��malar s�ras�nda diyabet �yk�s� olan veya olmayan, y�ksek v�cut kitle indekslerine (VK�) sahip hastalarda hiperglisemi bildirilmi�tir. Ancak, bir hiperglisemi olay� ya�ayan t�m hastalar serum
glukoz d�zeylerini yak�ndan takip ettirmelidir. Gerekli durumlarda anti- diyabetik tedavi
uygulanmal�d�r.
�nf�zyon b�lgesi ektravazasyonu
�ntraven�z inf�zyon esnas�nda ekstravazasyon meydana gelmi�tir. Ekstravazasyon ihtimaline kar��, ilac�n uygulanmas� s�ras�nda inf�zyon b�lgesinde infiltrasyon olup olmad���n�n yak�ndan izlenmesi tavsiye edilir.
B�brek ve karaci�er yetmezli�i
B�brek ve karaci�er yetmezli�i olan hastalar ile s�n�rl� deneyim mevcuttur. Eldeki veriler MMAE klirensinin �iddetli b�brek yetmezli�i, karaci�er yetmezli�i ve d���k serum alb�min konsantrasyonlardan etkilenebilece�ine i�aret etmektedir (bkz. b�l�m 5.2)
CD30+ CTCL
Mikozis fungoides (MF) ve primer kutan�z anaplastik b�y�k h�creli lenfoma (pcALCL) hari� CD30+ CTCL alt tiplerinde tedavi etkisinin boyutu, y�ksek dereceli klinik kan�t bulunmad���ndan dolay� a��k de�ildir. ADCETR�S ile ger�ekle�tirilen iki adet tek kollu faz II �al��mas�nda, Sézary sendromu (SS), lenfomatoid pap�loz (LyP) ve mikstCTCL histolojik alt tiplerinde hastal�k aktivitesi g�sterilmi�tir. Bu veriler, etkililik ve g�venlili�in di�er CTCL CD30+ alt tiplerine uyarlanabilece�ini �ne s�rmektedir. Bununla birlikte ADCETR�S, di�er CD30+ CTCL hastalar�nda bireye dayal� potansiyel yarar-risk oran�n�n dikkatli de�erlendirilmesinden sonra dikkatli �ekilde kullan�lmal�d�r (bkz. b�l�m 5.1).
Yard�mc� maddeler
Bu t�bbi �r�n her “doz”unda 1 mmol (23 mg)'dan daha az sodyum ihtiva eder; yani asl�nda “sodyum i�ermez”.
Takip edilebilirlik
Biyoteknolojik �r�nlerin takip edilebilirli�inin sa�lanmas� i�in uygulanan �r�n�n ticari ismi ve seri numaras� mutlaka hasta dosyas�na kaydedilmelidir.
4.5. Di�er t�bbi �r�nler ile etkile�imler ve di�er etkile�im �ekilleri
CYP3A4 yoluyla metabolize olan t�bbi �r�nler ile etkile�im (CYP3A4 inhibit�rleri/ind�kleyicileri)
ADCETR�S'in kuvvetli bir CYP3A4 ve P-gp inhibit�r� olan ketokonazol ile birlikte uygulanmas� sonucunda antimikrot�b�l ajan MMAE'ye maruziyet yakla��k %73 artm��t�r ve ADCETR�S'e plazma maruziyeti etkilenmemi�tir. Dolay�s�yla, ADCETR�S'in kuvvetli CYP3A4 ve P-gp inhibit�rleri ile bir arada uygulanmas�, n�tropeni insidans�n� art�rabilir. N�tropeni geli�ti�i takdirde Tablo 1 ve Tablo 2'de yer alan n�tropeni i�in doz uygulamas� �nerilerine ba�vurunuz (bkz. b�l�m 4.2).
ADCETR�S'in kuvvetli bir CYP3A4 ind�kleyicisi olan rifampisin ile birlikte uygulanmas�, ADCETR�S'e plazma maruziyetini etkilememi�tir. Farmakokinetik veriler s�n�rl� da olsa, rifampisin
ile birlikte kullan�m�n�n, test edilebilen MMAE metabolitlerinin plazma konsantrasyonlar�n� d���rd��� g�r�lm��t�r.
Bir CYP3A4 substrat� olan midazolam�n ADCETR�S ile birlikte uygulanmas�, midazolam metabolizmas�n� de�i�tirmemi�tir; dolay�s�yla, ADCETR�S'in, CYP3A4 enzimleri taraf�ndan metabolize olan ila�lara maruziyeti de�i�tirmesi beklenmez.
Doksorubisin, vinblastin ve dakarbazin (AVD)
Brentuximab vedotinin AVD ile kombinasyon �eklinde uygulanmas�n� takiben antikor ila� konjugat� (ADC; antibody drug conjugate) ve MMAE'nin s�ras�yla serum ve plazma farmakokinetik �zellikleri monoterapinin farmakokinetik �zelliklerine benzerdir.
Brentuximab vedotin ile e� zamanl� olarak verilmesi AVD'nin plazma seviyelerini etkilememi�tir. Siklofosfamid, Doksorubisin ve Prednizon (CHP)
CHP ile birlikte brentuximab vedotinin uygulanmas�n� takiben, s�ras�yla ADC ve MMAE'nin serum ve plazma farmakokinetik �zellikleri, monoterapideki ile benzerdir.
E� zamanl� brentuximab vedotin uygulamas�n�n CHP maruziyetini etkilemesi beklenmemektedir.
Bleomisin
Brentuximab vedotin ve bleomisin (B) ile formal ila�-ila� etkile�im �al��malar� yap�lmam��t�r. Bir faz I doz belirleme ve g�venlilik �al��mas�nda (SGN35-009) brentuximab vedotin art� ABVD ile tedavi edilen 25 hastadan 11'inde (%44) kabul edilemez pulmoner toksisite (2 fatal olay dahil) g�zlemlenmi�tir. Brentuximab vedotin art� AVD ile pulmoner toksisite ya da fatal olay bildirilmemi�tir. Dolay�s�yla ADCETR�S ile bleomisinin e� zamanl� kullan�lmas� kontrendikedir (bak�n�z b�l�m 4.3).
�zel pop�lasyonlara ili�kin ek bilgiler
Herhangi bir etkile�im �al��mas� yap�lmam��t�r.
Pediyatrik pop�lasyon
Herhangi bir etkile�im �al��mas� yap�lmam��t�r.
4.6. Gebelik ve laktasyon
Gebelik kategorisi: D
�ocuk do�urma potansiyeli bulunan kad�nlar / Do�um kontrol� (Kontrasepsiyon)
Gebe kalma potansiyeli olan kad�nlar ADCETR�S tedavisi s�ras�nda ve tedaviden sonra 6 aya kadar iki adet etkili kontraseptif y�ntem kullanmal�d�r.
Gebelik d�nemi
Gebe kad�nlarda ADCETR�S kullan�m�na ili�kin veri bulunmamaktad�r. Hayvanlar ile y�r�t�len �al��malar �reme toksisitesi g�stermi�tir (bkz. b�l�m 5.3).
ADCETR�S, anneye olan fayda fet�se olan potansiyel riske a��r basmad��� s�rece gebelik s�resince kullan�lma mal�d�r. E�er gebe bir kad�n�n tedavi edilmesi gerekli ise, anne aday� fet�s �zerindeki potansiyel risk konusunda a��k�a bilgilendirilmelidir.
ADCETR�S ile tedavi g�ren erkek partnerinden �ocuk sahibi olmak isteyen kad�nlara ili�kin tavsiyeler i�in a�a��daki �reme yetene�i/fertilite b�l�m�ne bak�n�z.
Laktasyon d�nemi
ADCETR�S/metabolitlerinin hayvan veya insan s�t�yle at�l�m�na ili�kin yeterli bilgi
bulunmamaktad�r.
Emzirilen bebekler i�in risk olas�l�k d��� b�rak�lamamaktad�r.
Emzirmenin bebe�e olan potansiyel riski ve tedavinin anneye olan faydas� g�z �n�nde bulundurularak emzirmenin sona erdirilip erdirilmeyece�i veya ADCETR�S tedavisinin sona erdirilip erdirilmeyece�i/ tedaviden ka��n�l�p ka��n�lmayaca�� y�n�nde bir karar verilmelidir.
�reme yetene�i /Fertilite
Klinik d��� �al��malara g�re ADCETR�S tedavisi, testik�ler toksisite ile sonu�lanabilir ve erkek fertilitesini de�i�tirebilir. MMAE'nin an�jenik �zellikleri oldu�u g�sterilmi�tir (bkz. b�l�m 5.3). Bu nedenle, bu ila� ile tedavi edilen erkeklerin tedaviden �nce spermlerinin dondurulmas� ve saklanmas� �nerilmektedir. Bu ila� ile tedavi edilen erkeklerin tedavi s�resinde ve son dozdan itibaren 6 aya kadar �ocuk sahibi olmamalar� tavsiye edilir.
4.7. Ara� ve makine kullan�m� �zerindeki etkiler
ADCETR�S'in ara� ve makine kullanma becerisi �zerinde orta derecede etkileri olabilir (�rn: ba� d�nmesi). Bkz: b�l�m 4.8.
4.8. �stenmeyen etkiler
G�venlilik profilinin �zeti
ADCETR�S'in g�venlilik profili mevcut klinik �al��ma verilerine, Tan�ml� Hasta Program�na (THP) ve g�n�m�ze kadar edinilmi� olan pazarlama sonras� deneyime dayanmaktad�r. A�a��da ve Tablo 5'te tan�mlanan advers reaksiyonlar�n s�kl�klar�, klinik �al��malarda elde edilen verilere dayal� olarak belirlenmi�tir.
Monoterapi
HL, sALCL ve CTCL �al��malar�nda (SG035-0003, SG035-0004, SGN35-005, SGN35-006,
C25001, C25006 ve C25007, bkz. b�l�m 5.1) monoterapi olarak ADCETR�S'in havuzlanm�� veri
setinde en s�k g�r�len advers olaylar (≥ %10) enfeksiyonlar, periferik duyusal n�ropati, bulant�, bitkinlik, diyare, pireksi, n�tropeni, �st solunum yolu enfeksiyonu, artralji, d�k�nt�, �ks�r�k, kusma, pir�rit, periferik motor n�ropati, inf�zyona ba�l� reaksiyonlar, konstipasyon, dispne, miyalji kilo kayb� ve abdominal a�r� olmu�tur.
Ciddi advers reaksiyonlar hastalar�n %12'sinde g�zlendi. Nadir g�r�len ciddi advers ila� reaksiyonlar�n�n s�kl��� < %1'dir.
Advers olaylar ADCETR�S alan hastalar�n %24'�n�n tedaviyi b�rakmas�na neden oldu.
ADCETR�S (SGN35-006, bkz. b�l�m 5.1) ile tekrar tedavi edilen hastalardaki g�venlilik verileri, daha y�ksek g�r�lme s�kl���na sahip (pivotal faz 2 �al��malar�nda %28 vs. %9) ve esas olarak derece 2 olan periferik motor n�ropati hari� kombine edilmi� pivotal faz 2 �al��malar�nda g�zlemlenenler ile tutarl� olmu�tur. Ayr�ca bu hastalarda artralji, derece 3 anemi ve s�rt a�r�s�, kombine edilmi� pivotal faz 2 �al��malarda g�zlenen hastalara k�yasla daha s�k g�r�lm��t�r.
�nerilen doz olan �� haftada bir 1,8 mg/kg ile tedavi edilen, otolog k�k h�cre nakli yap�lmam�� n�kseden veya tedaviye diren�li HL hastalar�ndaki g�venlik profili, tek kollu bir faz 4 �al��mada (n=60), faz 1 doz eskalasyon ve klinik farmakaloji �al��malar�nda (n=15 hasta) ve Tan�ml� Hasta Program�nda (THP) (n=26) (bkz. b�l�m 5.1), pivotal klinik �al��malardaki g�venlilik profili ile tutarl�d�r.
Kombinasyon tedavisi
ADCETR�S ile kombine verilen kemoterapi ila�lar� (doksorubisin, vinblastin ve dakarbazin (AVD) veya siklofosfamid, doksorubisin ve prednizon (CHP)) hakk�nda g�venlilik bilgileri i�in bu �r�nlerin k�sa �r�n bilgilerine ba�vurunuz.
Daha �nce tedavi edilmemi� ilerlemi� HL (C25003) g�r�len 662 hastada ve daha �nce tedavi edilmemi� CD30+ PTCL (SGN35-014) g�r�len 223 hastada kombinasyon tedavisi olarak ADCETR�S'in uyguland��� �al��malarda, en yayg�n advers reaksiyonlar (≥% 10) �unlar olmu�tur: enfeksiyonlar, n�tropeni, periferik duyusal n�ropati, bulant�, kab�zl�k, kusma, ishal, yorgunluk, pireksi, alopesi, anemi, kilo kayb�, stomatit, febril n�tropeni, kar�n a�r�s�, i�tah azalmas�, insomnia, kemik a�r�s�, d�k�nt�, �ks�r�k, dispne, artralji, miyalji, s�rt a�r�s�, periferik motor n�ropati, �st solunum yolu enfeksiyonu ve ba� d�nmesi.
ADCETR�S kombinasyon tedavisi alan hastalarda ciddi advers reaksiyonlar hastalar�n %34'�nde geli�mi�tir. Hastalar�n ≥ %3'�nde olu�an ciddi advers reaksiyonlar aras�nda febril n�tropeni (%15), ate� (%5) ve n�tropeni (%3) vard�r.
Advers olaylar hastalar�n %10'unda tedavinin b�rak�lmas�na neden olmu�tur. Hastalar�n ≥%2'sinde tedavinin b�rak�lmas�na yol a�an advers olaylar aras�nda periferik duyusal n�ropati ve periferik n�ropati vard�r.
Advers reaksiyonlar�n tablo halinde listesi
ADCETR�S i�in advers reaksiyonlar MedDRA Sistem Organ S�n�flamas� ve Tercih Edilen Terimler'e g�re listelenmi�tir. Her bir sistem organ s�n�f� i�erisinde advers reaksiyonlar s�kl�k kategorilerine g�re listelenmi�tir: �ok yayg�n (≥1/10), yayg�n (≥1/100 ila <1/10); yayg�n olmayan (≥1/1.000 ila 1/100); seyrek (≥1/10.000 ila 1/1.000); �ok seyrek (≤1/10.000), bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor). Her bir s�kl�k grubundaki advers reaksiyonlar, en ciddiden en hafife do�ru s�ralanm��t�r.
Tablo 5: ADCETR�S i�in advers reaksiyonlar
Sistem organ s�n�f� | Advers reaksiyonlar (monoterapi) | Advers reaksiyonlar (kombinasyon tedavisi) |
Enfeksiyonlar ve enfestasyonlar |
| |
�ok yayg�n: | Enfeksiyon, �st solunum yolu enfeksiyonu | Enfeksiyon, �st solunum yolu enfeksiyonu |
Yayg�n: | Herpes zoster, pn�moni, herpes simplex, oralkandidiyaz | Pn�moni, oral kandidiyaz, sepsis/septik �ok, herpes zoster |
Yayg�n olmayan: | Pneumocystis jiroveci pn�monisi, stafilokokal bakteriyemi, sitomegalovir�s enfeksiyonu veya reaktivasyonu, sepsis/septik �ok | Herpes simplex, Pneumocystis jiroveci pn�monisi |
Bilinmiyor: | Progresif multifokal l�koensefalopati |
|
Kan ve lenf sistemi hastal�klar� | ||
�ok yayg�n: | N�tropeni | N�tropeni, anemi, febril n�tropeni |
Yayg�n: | Anemi, trombositopeni | Trombositopeni |
Yayg�n olmayan: | Febril n�tropeni |
|
Ba����kl�k sistemi hastal�klar� | ||
Yayg�n olmayan: | Anafilaktik reaksiyon | Anafilaktik reaksiyon |
Metabolizma ve beslenme bozukluklar� | ||
�ok yayg�n |
| ��tahta azalma |
Yayg�n | Hiperglisemi | Hiperglisemi |
Yayg�n olmayan: | T�m�r lizis sendromu | T�m�r lizis sendromu |
Psikiyatrik hastal�klar | ||
�ok yayg�n |
| �nsomnia |
Sinir sistemi hastal�klar� | ||
�ok yayg�n: | Periferik duyusal n�ropati, periferik motor n�ropatisi | Periferik duyusal n�ropati, periferik motor n�ropatisi, sersemlik |
Yayg�n: | Sersemlik |
|
Yayg�n olmayan: | Demiyelinizan polin�ropati |
|
Solunum, g���s bozukluklar� ve mediastinal hastal�klar | ||
�ok yayg�n: | �ks�r�k, dispne | �ks�r�k, dispne |
Gastrointestinal hastal�klar |
|
|
�ok yayg�n: | Bulant�, diyare, kusma, konstipasyon, abdominal a�r� | Bulant�, konstipasyon, kusma, diyare, abdominal a�r�, stomatit |
Yayg�n olmayan: | Akut pankreatit | Akut pankreatit |
Hepatobiliyer hastal�klar | ||
Yayg�n: | Alanin aminotransferaz/aspartat aminotransferaz (ALT/AST) art��� | Alanin aminotransferaz/aspartat aminotransferaz (ALT/AST) art��� |
Deri ve deri alt� doku hastal�klar� | ||
�ok yayg�n: | D�k�nt�, pir�rit | Alopesi, d�k�nt� |
Yayg�n: | Alopesi | Pir�rit |
Yayg�n olmayan: | Stevens-Johnson sendromu/toksik epidermal nekroliz | Stevens-Johnson sendromu |
Bilinmiyor: | Eozinofili ve sistemik semptomlu ila� reaksiyonlar� (DRESS) |
|
Kas-iskelet bozukluklar�, ba� doku ve kemik hastal�klar� | ||
�ok yayg�n: | Artralji, miyalji | Kemik a�r�s�, artralji, miyalji, s�rt a�r�s� |
Yayg�n: | S�rt a�r�s� |
|
Genel bozukluklar ve uygulama b�lgesine ili�kin hastal�klar | ||
�ok yayg�n: | Yorgunluk, pireksi, inf�zyona ba�l� reaksiyonlara | Yorgunluk, pireksi |
Yayg�n: | Titreme | �nf�zyona ba�l� reaksiyonlara, titreme |
Bilinmiyor: | �nf�zyon b�lgesi ekstravazasyonu |
|
Ara�t�rmalar | ||
�ok yayg�n: | Kilo kayb� | Kilo kayb� |
aOrtak tercih edilen terminolojiyi temsil eder.
bToksik epidermal nekroliz, kombinasyon tedavisinde rapor edilmemi�tir.
c Ekstravazasyonla ilgili reaksiyonlara inf�zyon b�lgesinde deride k�zar�kl�k, a�r�, �i�me, kabarc�klanma, pul pul d�k�lme ve sel�lit dahildir.
Se�ili advers olaylar�n tan�m�
N�tropeni ve febril n�tropeni
Monoterapi
Klinik �al��malarda, n�tropeni hastalar�n %13'�nde doz ertelemelerine neden olmu�tur. Derece 3 n�tropeni hastalar�n�n %13'�nde ve derece 4 n�tropeni hastalar�n�n %5'inde g�r�lm��t�r. 1 hastada dozun azalt�lmas� ve 1 hastada n�tropeni tedavisinin b�rak�lmas� gerekmi�tir.
Bu tedavi ile �iddetli ve uzun s�reli (≥1 hafta) n�tropeni ortaya ��kabilir ve bu da hastalarda ciddi enfeksiyonlar�n geli�me riskini art�r�r. Febril n�tropeni hastalar�n <%1'inde rapor edilmi�tir (bkz. b�l�m 4.2).
Pivotal faz 2 pop�lasyonunda (SG035-0003 ve SG035-0004), Derece 3 veya Derece 4 n�tropeninin medyan s�resi s�n�rl�d�r (1 hafta); hastalar�n %2'sinde ≥ 7 g�n s�ren Derece 4 n�tropeni g�r�lm��t�r. Pivotal faz 2 pop�lasyonunda Derece 3 veya Derece 4 n�tropenisi olan hastalar�n yar�s�ndan az�nda zamansal ili�kili enfeksiyonlar belirtilmi� ve zamansal ili�kili enfeksiyonlar�n b�y�k k�sm� Derece 1 veya Derece 2 olmu�tur.
Kombinasyon tedavisi
Kombinasyon tedavisi olarak ADCETR�S ile yap�lan klinik �al��malarda n�tropeni, hastalar�n
%19'unda dozun verilmesinde gecikmelere yol a�m��t�r. Derece 3 n�tropeni, hastalar�n %17'sinde bildirilirken Derece 4 n�tropeni, hastalar�n %41'inde bildirilmi�tir. Hastalar�n y�zde ikisinde dozun azalt�lmas� gerekmi�tir ve <%1'i n�tropeni nedeniyle bir ya da daha fazla �al��ma ilac�n� b�rakm��t�r.
Febril n�tropeni G-CSF ile primer profilaksi almayan hastalar�n %20'sinde bildirilmi�tir (bak�n�z b�l�m 4.2). G-CSF ile primer profilaksi alan hastalarda febril n�tropeni s�kl��� %13 olarak bildirilmi�tir.
Ciddi enfeksiyonlar ve f�rsat�� enfeksiyonlar
Monoterapi
Klinik �al��malarda hastalar�n %10'unda ciddi ve f�rsat�� enfeksiyonlar g�r�lm��t�r, hastalar�n
%1'inden az�nda sepsis ve septik �ok g�r�lm��t�r. En s�k bildirilen f�rsat�� enfeksiyonlar herpes zoster ve herpes simplex olmu�tur.
Kombinasyon tedavisi
Kombinasyon tedavisi olarak ADCETR�S ile yap�lan klinik �al��malarda f�rsat�� enfeksiyonlar dâhil ciddi enfeksiyonlar hastalar�n %15'inde g�r�lm��t�r; sepsis, n�tropenik sepsis, septik �ok ya da bakteriyemi hastalar�n %4'�nde g�r�lm��t�r. En s�k bildirilen f�rsat�� enfeksiyonlar Herpes vir�s enfeksiyonlar�d�r.
Periferik n�ropati
Monoterapi
Klinik �al��malarda pop�lasyonun %57'sinda tedavi s�ras�nda geli�en n�ropati ve %13'�nde periferik motor n�ropati ortaya ��km��t�r. Periferik n�ropati hastalar�n %15'inde tedaviyi b�rakmaya, %15'inde
doz azalt�lmas�na ve %16's�nde doz ertelemesine neden olmu�tur. Periferik n�ropati g�r�len hastalarda periferik n�ropatinin ilk ba�lad��� tarihten itibaren ge�en medyan s�re 12 haftad�r.Periferik n�ropatiye ba�l� olarak tedaviyi b�rakan hastalar i�in medyan s�re 11 siklus olarak rapor edilmi�tir.
Pivotal faz 2 (SG035-0003 ve SG035-0004) �al��malar�nda ve randomize faz 3 monoterapi �al��malar�nda (SGN35-005 ve C25001) periferik n�ropati deneyimleyen hastalar aras�nda tedavi sonu ile son de�erlendirme aras�ndaki medyan s�re yakla��k 48,9 hafta ile 98 haftad�r. Son de�erlendirmenin yap�ld��� zamanda periferik n�ropati deneyimleyen hastalar�n �o�unda (%82-85) periferik n�ropati semptomlar�nda iyile�me ya da d�zelme olmu�tur. T�m olaylarda iyile�me ya da d�zelmenin ilk ba�lad��� tarihten itibaren ge�en medyan s�re 16 hafta ile 23,4 hafta aras�ndad�r.
ADCETR�S ile yeniden tedavi g�ren, n�kseden ve tedaviye diren�li klasik Hodgkin lenfoma ve sistemik anaplastik b�y�k h�creli lenfoma hastalar�nda (SGN35-006), son de�erlendirmede hastalar�n �o�unlu�unda (%80) periferik n�ropati semptomlar�nda gerileme veya iyile�me g�zlenmi�tir.
Kombinasyon tedavisi
AVD ile kombinasyon tedavisi olarak ADCETR�S ile yap�lan klinik �al��mada tedavi ile ortaya ��kan n�ropati pop�lasyonun %67'sinde g�r�lm��t�r; periferik motor n�ropati hastalar�n %11'inde geli�mi�tir. Periferik n�ropati hastalar�n %7'sinde tedavinin b�rak�lmas�na, %21'inde dozun azalt�lmas�na ve hastalar�n %1'inde dozun ertelenmesine yol a�m��t�r. Periferik n�ropati geli�en hastalarda periferik n�ropatinin ba�lamas�na kadar ge�en ortanca s�re 8 haftad�r. Periferik n�ropatiye ba�l� olarak tedaviyi b�rakan hastalar bir ya da daha fazla ila� kesilmeden �nce ortanca 8 doz ADCETR�S+AVD(A+AVD) alm��t�r.
Periferik n�ropati geli�en hastalarda tedavinin sonlanmas�nda son de�erlendirmeye kadar ge�en ortanca takip s�resi 91 haftad�r. Son de�erlendirme s�ras�nda hastalar�n �o�unlu�unda (%76) periferik n�ropati d�zelmi� ya da semptomlar�nda iyile�me olmu�tur. Ba�lang�c�ndan periferik n�ropati olaylar�n�n d�zelmesine ya da iyile�me g�stermesine kadar ge�en medyan s�re 10 hafta olmu�tur (0 hafta ile 139 hafta aras�nda de�i�mi�tir).
CHP ile kombinasyon tedavisi olarak ADCETR�S'in uyguland��� klinik �al��mada, pop�lasyonun
%52'sinde tedaviyle ortaya ��kan n�ropati g�r�l�rken hastalar�n %9'unda periferik motor n�ropati izlenmi�tir. Periferik n�ropati hastalar�n %1'inde tedavinin kesilmesine, hastalar�n %7'sinde dozun azalt�lmas�na ve hastalar�n <% 1'inde doz gecikmelerine yol a�m��t�r.
Periferik n�ropati g�r�len hastalarda medyan ba�lang�� s�resi 9,1 hafta olarak bulunmu�tur. Periferik n�ropati nedeniyle tedavinin kesildi�i hastalar, bir veya daha fazla ajan�n kesilmesinden �nce medyan 5 doz ADCETR�S + CHP (A+CHP) alm��t�r.
Periferik n�ropati g�r�len hastalar aras�nda tedavinin bitiminden son de�erlendirmeye kadar medyan takip s�resi yakla��k 177 hafta olmu�tur. Son de�erlendirme s�ras�nda, periferik n�ropati g�r�len hastalar�n % 64'�nde periferik n�ropati semptomlar� d�zelmi� veya iyile�me g�stermi�tir. Periferik n�ropati olaylar�n�n ba�lang�c�ndan olaylar�n d�zelmesine veya iyile�mesine kadar ge�en medyan s�re 19,0 hafta (0 hafta ile 205 hafta aras�nda de�i�mi�tir) olmu�tur.
�nf�zyona ba�l� reaksiyonlar
Monoterapi
Hastalar�n %12'sinde ba� a�r�s�, d�k�nt�, s�rt a�r�s�, kusma, titreme, bulant�, dispne, ka��nt� ve �ks�r�k gibi inf�zyona ba�l� reaksiyonlar bildirilmi�tir.
Anafilaktik reaksiyonlar bildirilmi�tir (bkz. b�l�m 4.4). Bir anafilaktik reaksiyonun semptomlar�, bunlarla s�n�rl� olmamak �zere �rtiker, anjiyo�dem, hipotansiyon ve bronkospazm� i�erir.
Kombinasyon tedavisi
Ba� a�r�s�, d�k�nt�, s�rt a�r�s�, kusma, titremeler, bulant�, dispne, ka��nt�, �ks�r�k, inf�zyon yerinde a�r� ve ate� gibi IRR'ler hastalar�n %8'inde bildirilmi�tir. Anafilaktik reaksiyonlar bildirilmi�tir (bak�n�z b�l�m 4.4). Anafilaktik reaksiyon semptomlar� aras�nda bunlarla k�s�tl� olmamakla birlikte �rtiker, anjiyo�dem, hipotansiyon ve bronkospazm vard�r.
�mm�nojenisite
Klinik �al��malarda, hastalar periyodik olarak duyarl� bir elektrokemiluminesan immunoassay y�ntemi kullanarak ADCETR�S antikorlar� a��s�ndan test edildi. Anl�k olarak pozitif veya negatif test sonu�lar� elde edilen hastalarla k�yasland���nda ADCETR�S antikorlar� ile tedavi g�ren hastalarda inf�zyona ba�l� reaksiyonlarda y�ksek insidans g�r�lm��t�r.
ADCETR�S antikorlar�n�n varl���, serum ADCETR�S d�zeylerinde klinik a��dan anlaml� azalmalar ile korelasyon g�stermemi�tir ve ADCETR�S'in etkilili�inde azalmaya neden olmam��t�r. ADCETR�S'e y�nelik antikorlar�n varl���, kesin olarak bir inf�zyona ba�l� reaksiyonun geli�imini �ng�rmezken; ge�ici olarak pozitif anti ila� antikorlar� (A�A) olan hastalarla ve hi�bir zaman A�A pozitif olmayan hastalarla kar��la�t�r�ld���nda , s�rekli pozitif anti ila� antikorlar� olan hastalarda inf�zyona ba�l� reaksiyonlar�n insidans� daha y�kse k olmu�tur.
Monoterapi �al��mas� C25002
A�A pozitif oldu�u teyit edilen pediyatrik hastalarda ADCETR�S'in klirensinde artma trendi g�r�lm��t�r. 12 ya��ndan k���k hi�bir hastada (11 hastan�n 0'�) ve 12 ya� ve �zerinde olan hastalar�n 2'sinde (23 hastan�n 2'si) A�A s�rekli pozitif olmu�tur.
Kombinasyon Kullan�m �al��mas� C25004
�al��ma C25004'de A�A pozitiflik oran� d���k olup; 59 hastadan (12 ya� ve �zeri) 4 hasta ge�ici olarak A�A pozitif olmu� ve hi�bir hasta kal�c� olarak A�A pozitif olmam��t�r. Ge�ici A�A pozitif hastalar�n say�s�n�n azl��� nedeniyle A�A'n�n etkililik �zerindeki etkisi sonu�suzdur.
Pediyatrik pop�lasyon
Monoterapi �al��mas� C25002
G�venlilik n�kseden veya tedaviye diren�li Hodgkin lenfoma ve sALCL olan 7-17 ya� aral���ndaki pediyatrik hastalarda (n=36) faz 1/2 �al��mas� ile de�erlendirilmi�tir (bkz. B�l�m 5.1.). Bu �al��madaki 36 hastada herhangi bir yeni g�venlik ��phesi rapor edilmemi�tir.
Kombinasyon Kullan�m �al��mas� C25004
�nceden tedavi edilmemi� ileri evre klasik CD30+ HL'l� 6-17 ya� aras� 59 pediyatrik hastada
kemoterapi ile kombinasyon halinde a��k etiketli, �ok merkezli bir �al��mada g�venlilik de�erlendirilmi�tir (Bkz b�l�m 5.1). Bu �al��mada herhangi bir yeni g�venlilik endi�esi bildirilmemi�tir. Bu �al��mada bildirilen en yayg�n ciddi advers reaksiyon febril n�tropenidir (%17). G-CSF profilaksisinin doktorun tercihine ba�l� oldu�u d���n�lm��t�r. Periferik n�tropati olaylar� (Standardize MedDRA sorgusuna g�re), bu �al��madaki pediyatrik hastalar�n %24'�nde bildirilmi�tir.
Geriyatrik pop�lasyon
Monoterapi
Ya�l� hastalardaki g�venlilik profili eri�kin hastalardaki profille uyumludur. Ancak ya�l� hastalar pn�moni, n�tropeni ve febril n�tropeni gibi olaylara daha fazla duyarl� olabilir.
Kombinasyon tedavisi
Ya�l� hastalarda (≥ 60 ya�; n = 186 [%21]), advers olaylar�n s�kl��� tedavi kollar� aras�nda benzerdir. Genel �al��ma pop�lasyonu ile kar��la�t�r�ld���nda ya�l� hastalarda daha ciddi advers olaylar ve doz de�i�iklikleri (doz ertelemeleri, azaltmalar� ve b�rak�lmas� dâhil) bildirilmi�tir. �leri ya� her iki kolda febril n�tropeni i�in bir risk fakt�r� olmu�tur. G-CSF primer profilaksisi alan ya�l� hastalarda G- CSF primer profilaksisi almayanlara g�re n�tropeni ve febril n�tropeni s�kl��� daha d���k olmu�tur.
��pheli advers reaksiyonlar�n raporlanmas�
Ruhsatland�rma sonras� ��pheli ila� advers reaksiyonlar�n�n raporlanmas� b�y�k �nem ta��maktad�r. Raporlama yap�lmas�, ilac�n yarar/risk dengesinin s�rekli olarak izlenme sine olanak sa�lar. Sa�l�k mesle�i mensuplar�n�n herhangi bir ��pheli advers reaksiyonu T�rkiye Farmakovijilans Merkezi (T�FAM)'ne bildirmeleri gerekmektedir (www.titck.gov.tr ; e-posta: tufam@titck.gov.tr; tel: 0 800 314 00 08; faks: 0 312 218 35 99)
4.9. Doz a��m� ve tedavisi
ADCETR�S doz a��m� i�in bilinen bir antidot yoktur. Doz a��m� durumunda hastalar ba�ta n�tropeni olmak �zere advers reaksiyonlara kar�� yak�ndan izlenmelidir ve destekleyici tedavi uygulanmal�d�r (bkz. b�l�m 4.4).
5. FARMAKOLOJ�K �ZELL�KLER
5.1. Farmakodinamik �zellikler
Farmakoterap�tik grup: Antineoplastik ajanlar; monoklonal antikorlar ve antikor ila� konjugatlar� ATC kodu: L01FX05
Etki mekanizmas�
ADCETR�S, bir antineoplastik ajan veren antikor ila� konj�gat�d�r (ADC); bu antineoplastik ajan, CD30 eksprese eden t�m�r h�crelerinde selektif olarak apoptotik h�cre �l�m�ne neden olur. Klinik d��� veriler, ADCETR�S'in biyolojik aktivitesinin �ok ad�ml� bir s�re�le meydana geldi�ini g�stermektedir. ADC'nin h�cre y�zeyinde CD30'a ba�lanmas�, ADC-CD30 kompleksinin internalizasyonunu ba�lat�r, bu daha sonra lizozomal kompartmana iletilir. H�cre i�inde tan�mlanm�� tek aktif par�a olan MMAE, proteolitik ayr�lma ile sal�n�r. MMAE'nin t�b�line ba�lanmas�, h�cre i�indeki mikrot�b�l a��n� bozar, h�cre d�ng�s�n�n durmas�n� ind�kler ve CD30 eksprese eden t�m�r h�crelerin apoptotik h�cre �l�m�ne neden olur.
Klasik Hodgkin lenfoma, sistemik anaplastik b�y�k h�creli lenfoma vek�tan�z t h�creli lenfomalar�n altt�rleri [Mikozis fungoides (MF) ve primer kutan�z anaplastik b�y�k h�creli lenfoma (pcALCL) dahil] CD30'u antijen olarak malign h�crelerinin y�zeyinde eksprese ederler. Bu ekspresyon hastal���n evresinden, tedavinin basama��ndan ve nakil durumundan ba��ms�zd�r. Bu �zellikler CD30'u terap�tik m�dahale i�in hedef k�lar. CD30 hedefli etki mekanizmas� nedeniyle ADCETR�S, kemo-direnci a�abilir ��nk� �oklu ajanl� kemoterapiye diren�li olan hastalarda CD30, �nceki nakil durumu fark etmeksizin s�rekli olarak eksprese olur. ADCETR�S'in CD30 hedefli mekanizmas�, klasik Hodgkin lenfoma, sistemik anaplastik b�y�k h�creli lenfoma ve CD30+ k�tan�z T h�creli lenfomalarda CD30'un tutarl� ekspresyonu, tedavi spektrumlar� ve CD30 pozitif malignitelerdeki �oklu tedavi serilerini takip eden klinik bulgular, ADCETR�S'in otolog k�k h�cre transplant� olsun veya olmas�n n�kseden veya tedaviye diren�li klasik HL'de, sALCL'de ve CD30 + CTCL hastalar�nda en az 1 seri sistemik tedavi sonras� kullan�m�na y�nelik biyolojik kan�t sa�lar.
Etki mekanizmas�na di�er antikorlarla ili�kili fonksiyonlar�n katk�s� d��lanmam��t�r. Farmakodinamik etkiler
Kardiyak elektrofizyoloji
Faz 1, tek kollu, a��k etiketli, �ok merkezli kardiyak g�venlilik �al��mas� kapsam�nda 3 haftada bir 1,8 mg/kg ADCETR�S ile tedavi edilen 52 hastadan CD30 eksprese eden hematolojik maligniteleri olan k�rk alt� (46) hasta de�erlendirilmi�tir. Birincil ama�, ADCETR�S'in kardiyak ventrik�ler re- polarizasyon �zerindeki etkisi de�erlendirmek olmu� ve �nceden tan�mlanm�� olan birincil analiz, QTc de�erinde ba�lang��tan, 1. siklustaki �oklu zaman noktalar� aras�nda de�i�ikli�i i�ermi�tir.
QTc �zerindeki etkinin �st %90 g�ven aral��� (GA) Siklus 1 ve Siklus 3 ba�lang�� sonras� zaman noktalar�n�n her biri i�in <10 milisaniye olmu�tur. Bu veriler, CD30 eksprese eden maligniteleri olan hastalarda 3 haftada bir 1,8 mg/kg dozunda uygulanan ADCETR�S'e ba�l� olarak klinik olarak anlaml� QT uzamas�n�n olmad���na i�aret etmektedir.
Klinik etkililik ve g�venlilik
Hodgkin lenfoma
�al��ma C25003
ADCETR�S'in kemoterapi ile kombinasyonunun (doksorubisin [A], vinblastin [V] ve dakarbazin [D] [AVD]) etkilili�i ve g�venlili�i daha �nce tedavi g�rmemi� 1334 ileri evre HL hastas�nda randomize, a��k etiketli, 2 kollu, �ok merkezli bir �al��mada de�erlendirilmi�tir. T�m hastalarda histolojik olarak do�rulanm�� CD30 eksprese eden hastal�k bulunmaktad�r. Hastalar�n y�zde altm�� ikisinde ekstranodal tutulum saptanm��t�r. 1334 hastadan 664'� ADCETR�S + AVD koluna ve 670 hasta ABVD (doksorubisin [A], bleomisin [B], vinblastin [V] ve dakarbazin [D]) koluna randomize edilmi� ve Uluslararas� Prognostik Fakt�r Projesi (IPFP) risk fakt�rleri ve b�lgeye g�re katmanlara ayr�lm��t�. Hastalar her 28 g�nl�k k�r�n 1. ve 15. G�n�nde 30 dakikal�k intraven�z inf�zyonla 1,2 mg/kg ADCETR�S + doksorubisin 25 mg/m², vinblastin 6 mg/m²ve dakarbazin 375 mg/m²alm��t�r. Al�nan ortanca k�r say�s� alt�d�r (aral�k: 1 - 6 k�r). Tablo 6 ba�lang��taki hasta ve hastal�k �zelliklerini vermektedir. �ki kol aras�nda hasta ve hastal�k �zellikleri a��s�ndan anlaml� farklar bulunmamaktad�r.
Tablo 6: Faz 3 daha �nce tedavi edilmemi� HL �al��mas�nda ba�lang��taki hasta ve hastal�k �zelliklerinin �zeti
Hasta �zellikleri | ADCETR�S + AVD n = 664 | ABVD n = 670 |
Ortanca ya� (aral�k) | 35 ya� (18-82) | 37 ya� (18-83) |
≥ 65 ya� hastalar n (%) | 60 (9) | 62 (9) |
Cinsiyet, n (%) | 378E (57) 286K (43) | 398E (59) 272K (41) |
ECOG durumu, n (%) |
|
|
0 | 376 (57) | 378 (57) |
1 | 260 (39) | 263 (39) |
2 | 28 (4) | 27 (4) |
Eksik | 0 | 2 |
Hastal�k �zellikleri |
|
|
HL tan�s�ndan ilk doza ortanca zaman (aral�k) | 0,92 ay (0,1-21,4) | 0,89 ay (0,0-81.4) |
�lk HL tan�s� konuldu�u s�rada hastal�k evresi, n (%) |
|
|
III | 237 (36) | 246 (37) |
IV | 425 (64) | 421 (63) |
Ge�erli de�il | 1 (< 1) | 1 (< 1) |
Eksik | 0 | 2 (<1 ) |
Tan� konuldu�u s�rada ekstranodal tutulum, n (%) | 411 (62) | 416 (62) |
IPFPrisk fakt�rleri, n (%) |
|
|
0-1 | 141 (21) | 141 (21) |
2-3 | 354 (53) | 351 (52) |
4-7 | 169 (25) | 178 (27) |
Tan� konuldu�u s�rada ya da �al��maya giri�te kemik ili�i tutulumu, n (%) | 147(22) | 151 (23) |
B semptomlar�a n (%) | 400 (60) | 381 (57) |
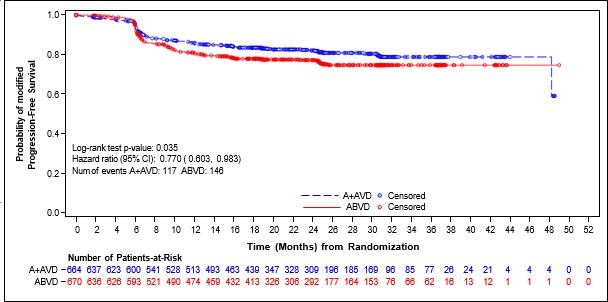
Randomizasyondan hastal�k progresyonuna, �l�me ya da ba��ms�z inceleme kurulu�u(IRF) de�erlendirmesine g�re birinci basamak tedaviden sonra tam olmayan yan�t (TY d���) bulgular�na ve takip eden kanser tedavisine kadar ge�en zaman olarak tan�mlanan, modifiye PFS (mPFS) C25003 �al��mas�nda birincil sonlan�m noktas�d�r. Modifiye olay�n zamanlamas� birinci basamak tedavinin tamamlanmas�ndan sonra Deauville skoru ≥3 olarak tan�mlanan tam yan�t�n (TY) yoklu�unu g�steren ilk PET incelemesidir. IRF de�erlendirmesine g�re medyan modifiye PFS'ye iki kolda da ula��lmam��t�r. Tedavi ama�lanan pop�lasyondaki (ITT) bulgular ADCETR�S+AVD i�in modifiye PFS'de istatistiksel olarak anlaml� iyile�me oldu�unu g�stermi�tir ve katmanl� tehlike oran� 0,770 (%95 GA, 0,603; 0,983, p = 0,035) olarak belirlenmi� olup ABVD ile kar��la�t�r�ld���nda
ADCETR�S+AVD i�in modifiye PFS olaylar� riskinde %23 azalma oldu�unu g�stermektedir.
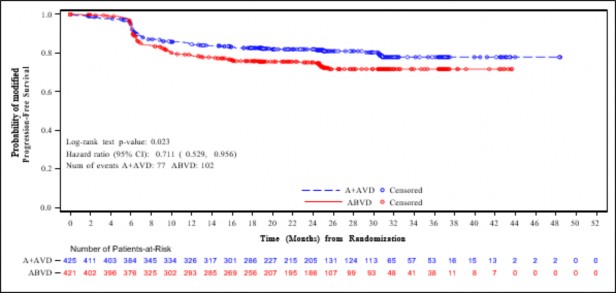
Hastal�k evresine g�re mPFS i�in yap�lan �nceden belirlenmi� bir alt grup analizi Evre IV hastal��� olan hastalarda etkinin ITT pop�lasyonuna g�re daha fazla oldu�unu g�stermi�tir ve katmanl� olmayan tehlike oran�n�n 0,71 (%95 GA, 0,53; 0,96) olmas� ABVD ile kar��la�t�r�ld���nda ADCETR�S+AVD i�in modifiye PFS olaylar� riskinde %29 azalma oldu�una i�aret etmektedir. ITT pop�lasyonunda 846 hastada (%64) Evre IV hastal�k saptanm��t�r.
Tablo 7 ITT pop�lasyonunda ve Evre IV hastal��� olan hastalarda modifiye PFS ve genel sa�kal�m (OS) i�in etkililik sonu�lar�n� vermektedir.
Tablo 7: 28 g�nl�k bir k�r�n 1. ve 15. G�n�nde 1,2 mg/kg ADCETR�S + AVD ile tedavi edilen daha �nce tedavi g�rmemis HL hastalar�nda etkililik sonu�lar� (ITT ve Evre IV)
| Tedavi Ama�lanan (ITT) Pop�lasyon | Evre IV Hastal��� Olan Hastalar | ||||
| ADCETR�S + AVD n = 664 | ABVD n = 670 | Katmanl� Tehlike Oran� ve p de�eri | ADCETR�S + AVD n = 425 | ABVD n = 421 | Katmanl� Olmayan Tehlike Oran� ve |
p de�eri | ||||||
Olay | 117 (18) | 146 (22) |
0,77 (%95 GA [0,60, 0,98]) p-de�eri =0,035 | 77 (18) | 102 (24) |
|
say�s� (%) | 0,71 | |||||
2. y�lda IRF'ye G�re Tahmini mPFS |
82,1 (%95 GA [78,8, 85,0]) |
77,2 (%95 GA [73,7, 80,4]) |
82,0 (%95 GA [77,8, 85,5]) |
75,3 (%95 GA [70,6, 79,3]) | (%95 GA [0,53, 0,96]) p-de�eri =0,023 | |
(%) | ||||||
Genel |
28 (4) |
39 (6) | 0,73 |
14 (3) |
26 (6) | 0,51 |
Sa�kal�m�l�m | (%95 GA [0,45, 1,18]) | (%95 GA [0,27, | ||||
Say�s� (%) | p-de�eri=0,199 | 0,97]) p-de�eri =0,037 | ||||
�ekil 1: ITT pop�lasyonunda IRF'ye g�re modifiye progresyonsuz sa�kal�m (ADCETR�S+ AVD ile ABVD kar��la�t�rmas�)
�ekil 2: Evre IV hastal��� olan hastalarda IRF'ye g�re modifiye progresyonsuz sa�kal�m (ADCETR�S + AVD ile ABVD kar��la�t�rmas�)
Randomizasyon rejiminin sonunda CR oran� ve ORR, birinci basamak tedavinin sonunda CR oran� ve 2. K�r�n sonunda PET negatifli�i oran�, yan�t s�resi (DOR), tam remisyon s�resi (DOCR), hastal�ks�z sa�kal�m (DFS) ve olays�z sa�kal�m (EFS) dâhil di�er ikincil etkililik noktalar�n�n hepsi hem ITT hem de Evre IV pop�lasyonunda ADCETR�S + AVD lehine e�ilim g�stermi�tir.
IRF'ye g�re modifiye PFS'nin �nceden belirlenmi� ya�, b�lge, ba�lang��taki kanser evresi, ba�lang��taki ekstranodal b�lgeler, IPFP risk fakt�rlerinin say�s�, ba�lang��taki B semptomlar�, 2.siklustaki PET de�erlendirmesi, 2.siklustaki PET Deauville skoru ve alternatif birinci basamak ila� (AFM) al�nmas� dâhil ITT alt grup analizleri yap�lm��t�r. Analizler alt gruplar�n �o�unda tutarl� bir �ekilde, ABVD ile kar��la�t�r�ld���nda ADCETR�S+AVD alan hastalar i�in yarar e�ilimi
g�stermi�tir. Ya�l� hasta pop�lasyonunda ( ≥ 60 ya� [n = 186] [HR = 1,00, %95 GA (0,58, 1,72)]
ve ≥ 65 ya� [n = 122] [HR = 1,01, %95 GA (0,53, 1,94)]) ve ekstranodal tutulumu olmayan hastalarda (n = 445) (HR = 1,04, %95 GA [0,67, 1,62]) yap�lan etkililik analizi iki kol aras�nda klinik olarak anlaml� fark olmad���n� g�stermi�tir.
Evre IV hastal��� olan hastalar i�in IRF'ye g�re modifiye PFS'nin ya�, b�lge, ba�lang��taki ekstranodal b�lgeler, IPFP risk fakt�rlerinin say�s�, ba�lang��taki B semptomlar�, ba�lang��taki ECOG durumu ve cinsiyet dahil post-hoc alt grup analizleri yap�lm��t�r. Analizler alt gruplar�n �o�unda tutarl� bir �ekilde ABVD ile kar��la�t�r�ld���nda ADCETR�S+AVD alan hastalar i�in yarar e�ilimi g�stermi�tir. Ekstranodal hastal���n bildirildi�i Evre IV hastal��� olan hastalarda ([n = 722] [HR = 0,69, %95 GA (0,50, 0,94)]) mPFS (IRF'ye g�re) yarar� g�sterilmi�tir. Ekstranodal hastal���n bildirilmedi�i Evre IV hastal��� olan hastalar i�in analizin yap�ld��� zaman itibariyle yarar g�sterilmemi�tir ([n = 85] [HR = 1,49, %95 GA (0,51, 4,31)]). Ekstranodal hastal��� olmayan Evre IV HL hastalar�ndaki bu bulgunun anlaml�l��� hasta say�s�n�n az olmas� ve d���k olay oranlar� (14 olay) nedeniyle belirlenememi�tir. A + AVD kolundaki (≥ 60 ya� [n = 118] [HR = 0,80, %95 GA (0,42, 1,53)] ve ≥ 65 ya� [n = 78] [HR = 0,78, %95 GA (0,36, 1,67) hastalar]) Evre IV hastal��� olan ya�l� hastalarda etkililik, ITT pop�lasyonundaki ya�l� hastalara oranla bu pop�lasyonda yarar�n daha b�y�k oldu�unu ortaya koymu�tur.
ITT pop�lasyonunda ADCETR�S+AVD ile tedavi edilen hastalar takip eden kurtarma kemoterapisi
ve y�ksek doz kemoterapi ve nakil a��s�ndan ABVD ile tedavi edilenlerle kar��la�t�r�ld�klar�nda %33 d���� g�zlenmi� olup ADCETR�S + AVD sonras� kurtarma kemoterapisi ya da y�ksek doz kemoterapi ve nakil tedavisi alanlar�n say�s� s�ras�yla n= 66 ven= 36 iken ABVD ile s�ras�yla n = 99 ve n = 54 olmu�tur. Evre IV hastalarda ADCETR�S + AVD sonras� kurtarma kemoterapisi alanlar (n=45) ABVD sonras� kurtarma kemoterapisi alanlara (n=69) g�re %35 daha d���kt�r ve kemoterapi ve nakil tedavisi alanlar�n say�s� ADCETR�S+AVD ile tedavi edilenlerde n=29 iken ABVD ile tedavi edilenlerde n=37 olup ADCETR�S + AVD ile tedavi edilenlerde %22 daha d���kt�r.
Avrupa Kanser Ara�t�rmas� ve Tedavisi �rg�t� Ya�am Kalitesi 30 maddeli Anketi (EORTC- QLQ- C30) hem ITT hem de Evre IV pop�lasyonunda iki kol aras�nda klinik olarak anlaml� fark olmad���n� g�stermi�tir.
�al��ma SGN35-005
ADCETR�S'in etkilili�i ve g�venlili�i ASCT'yi takiben relaps ya da progresyon riski alt�nda olan 329 HL hastas�nda randomize, �ift k�r, plasebo-kontroll�, 2 kollu �ok merkezli bir �al��mada de�erlendirilmi�tir. PML dahil bilinen serebral/meninjeal hastal��� olan hastalar �al��madan d��lanm��lard�r. Hasta �zellikleri i�in Tablo 8'e bak�n�z. 329 hastadan 165 hasta tedavi koluna randomize edilirken 164 hasta plasebo koluna randomize edilmi�tir. �al��mada hastalar�n ASCT'yi takiben iyile�tikten sonra (ASCT'yi takiben 30-45 g�n aras�nda) ilk dozlar�n� almalar� zorunlu tutulmu�tur. Hastalar 16 k�re kadar her 3 haftada bir 30 dakikada intraven�z olarak 1,8 mg/kg ADCETR�S ya da benzer g�r�n�ml� plasebo ile tedavi edilmi�tir.
�al��maya al�nmak i�in uygun hastalar�n a�a��da belirtilen risk fakt�rlerinden en az birine sahip olmalar� gerekmi�tir:
Birinci basamak tedaviye yan�ts�z HL
5.2. Farmakokinetik �zellikler
Genel �zellikler
Monoterapi
ADCETR�S'in farmakokinetik �zellikleri faz 1 �al��malarda ve 314 hastan�n verilerinin pop�lasyon farmakokineti�i analizinde de�erlendirilmi�tir. T�m klinik �al��malarda ADCETR�S, intraven�z inf�zyon �eklinde uygulanm��t�r.
ADCETR�S antikor ila� konjugat�n�n (A�K) maksimum konsantrasyonlar� tipik olarak inf�zyonun sonunda veya inf�zyonun sonuna en yak�n �rnek alma zaman� noktas�nda g�zlemlenmi�tir. A�K serum konsantrasyonlar�nda , yakla��k 4 ila 6 g�nl�k terminal yar� �m�r ile �ok �sl� bir d���� g�zlenmi�tir. Maruziyetlerin yakla��k olarak dozla oransal oldu�u g�r�lm��t�r. Tahmini terminal yar� �m�r ile uyumlu olarak, �� haftada bir tedavi program�nda A�K birikiminin minimal oldu�u ya da birikim olmad��� g�zlenmi�tir. Faz 1 �al��mada tek 1,8 mg/kg sonras�nda A�K'nin tipik Cve EAA de�erleri s�rs�yla yakla��k 31,98mcg/mLve 79,41mcg/mL x g�n bulunmu�tur.
MMAE, ADCETR�S'in ana metabolitidir. Faz 1 �al��mada A�K'nin tek 1,8 mg/kg'� sonras�nda MMAE'nin medyan C, EAA ve Tde�erlerinin s�ras�yla yakla��k 4,97 ng/mL, 37,03 ng/mL x g�n ve 2,09 g�n oldu�u belirlenmi�tir. MMAE maruziyetleri �oklu ADCETR�S dozlar�ndan sonra d��m�� olup sonraki dozlarda ilk dozun maruziyetinin yakla��k %50 ila %80'i g�zlenmi�tir. MMAE daha ileri metabolize olarak b�y�k �l��de e�it derecede g��l� bir metabolite d�n���r; ancak maruziyet derecesi MMAE'den daha d���k d�zeydedir. Bu nedenle MMAE'nin sistemik etkilerine �nemli bir katk�s� olmas� beklenmez.
�lk siklustaki daha y�ksek MMAE maruziyeti, n�trofil say�s�ndaki mutlak d���� ile ili�kili olmu�tur.
Kombinasyon tedavisi
ADCETR�S'in AVD ile kombine halde farmakokineti�i, 661 hastada tek bir �al��ma ile de�erlendirilmi�tir. Pop�lasyon farmakokinetik analizi, ADCETR�S'in AVD ile kombine halde farmakokineti�inin monoterapi ileuyumlu oldu�unu g�stermi�tir.
�ki haftada bir �oklu doz 1,2 mg/kg brentuximab vedotin intraven�z inf�zyonunu takiben inf�zyonun sonlar�na do�ru ADC'nin maksimum konsantrasyonuna eri�ilmi� ve eliminasyon multi- eksponansiyel bir azalma g�stererek tyakla��k 4-5 g�ne ula�m��t�r. �nf�zyonun bitmesinden yakla��k 2 g�n sonra 32 MMAE'lik maksimum plazma konsantrasyonlar�na eri�ilmi� ve mono- eksponansiyel bir azalma g�stererek tyakla��k 3-4 g�ne ula�m��t�r.
�ki haftada bir �oklu doz 1,2 mg/kg'l�k brentuximab vedotin intraven�z inf�zyonundan sonra 3. Siklusa kadar ADC ve MMAE kararl� durum vadi konsantrasyonuna ula��lm��t�r. Kararl� duruma ula��ld�ktan sonra, ADC'nin farmakokineti�i zamana ba�l� de�i�im g�stermemi�tir. ADC ak�m�lasyonu (1. ve 2. Sikluslar aras�nda AUCile hesaplanan) 1,27 kat olmu�tur. MMAE (1. ve 3. Sikluslar aras�nda AUCile hesaplanan), zamanla %50'ye yak�n azalma g�stermi�tir.
CHP ile kombinasyon halinde ADCETR�S'in farmakokineti�i, 223 hastada yap�lan tek fazl� bir �al��mada de�erlendirilmi�tir (SGN35-014). Her 3 haftada bir 1,8 mg / kg ADCETR�S'in �oklu doz
IV inf�zyonundan sonra, ADC ve MMAE'nin farmakokineti�i monoterapidekine benzer bulunmu�tur.
Emilim
ADCETR�S intraven�z yolla uygulan�r.
Da��l�m
�n vitro ortamda MMAE'nin insan serum plazma proteinine ba�lanma oran� %68-%82 aral���nda de�i�mi�tir. MMAE'nin y�ksek oranda proteine ba�lanan ila�lar�n yerini de�i�tirmesi ya da bu ila�lar taraf�ndan yerinin de�i�tirilmesi olas� de�ildir. �n vitro ko�ullarda MMAE'nin klinik konsantrasyonlarda P-gp substrat� oldu�u ve P-gp inhibit�r� olmad��� saptanm��t�r.
�nsanda ortalama kararl� durum da��l�m hacmi A�K i�in yakla��k 6-10 L bulunmu�tur. Pop�lasyon farmakokineti�i tahminlerine dayal� olarak, MMAE'nin tipik g�r�n�r santral da��l�m hacmi 35,5 L'dir.
Biyotransformasyon
A�K'nin protein olarak katabolize olmas�, komponent amino asitlerin geri d�n��mesi ya da elimine olmas� beklenir.
Hayvanlar ve insanlardaki in vivo veriler, ADCETR�S'ten sal�nan MMAE'nin sadece k���k bir k�sm�n�n metabolize oldu�unu g�stermektedir. MMAE metabolitlerinin d�zeyleri insan plazmas�nda �l��lmemi�tir. �n vitro ortamda MMAE'nin en az bir metabolitinin aktif oldu�u g�sterilmi�tir.
MMAE, CYP3A4'�n ve olas�l�kla CYP2D6'n�n substrat�d�r. �n vitro veriler MMAE metabolizmas�n�n temelde CYP3A4/5 ile oksidasyon yoluyla olu�tu�una i�aret etmektedir. �nsan karaci�eri mikrozomlar�n�n kullan�ld��� in vitro �al��malar ise MMAE'nin CYP3A4/5'i sadece klinik uygulama s�ras�nda elde edilmi� olan konsantrasyonlar�n �ok �zerinde ki konsantrasyonlarda inhibe etti�ini g�stermektedir. MMAE di�er izoformlar� inhibe etmez.
MMAE, insan hepatositlerinin primer k�lt�rlerinde ba�l�ca CYP450 enzimlerinin herhangi birini ind�klememi�tir.
Eliminasyon
A�K katabolizma yoluyla ve s�ras�yla 1,5 L/g�n ve 4-6 g�nl�k tipik olarak tahmini CL ve yar� �m�r ile elimine olur.
MMAE'nin eliminasyonu, A�K'den sal�nma h�z� ile s�n�rlanm��t�r; MMAE'nin tipik g�r�n�r CL ve yar� �m�r de�erleri s�ras�yla 19,99 L/g�n ve 3-4 g�n bulunmu�tur.
1,8 mg/kg ADCETR�S dozu alan hastalarda bir �trah �al��mas� ger�ekle�tirilmi�tir. ADCETR�S inf�zyonu s�ras�nda A�K i�eri�i olarak uygulanan toplam MMAE'nin yakla��k %24'� bir haftal�k s�rede hem idrar hem de fe�este tespit edilmi�tir. Tespit edilen MMAE'nin yakla��k %72'i fe�este bulunmu�tur. �drarda daha d���k miktarda MMAE (%28) at�lm��t�r.
Do�rusall�k/Do�rusal olmayan durum Veri yoktur.
�zel Pop�lasyonlar
Pop�lasyon farmakokineti�i analizi, ba�lang��taki serum alb�min konsantrasyonunun MMAE klirensi a��s�ndan �nemli bir e�de�i�ken oldu�unu g�stermi�tir. Bu analiz, serum alb�min konsantrasyonlar� normal aral�k i�erisinde olan hastalar ile kar��la�t�r�ld���nda serum alb�min konsantrasyonlar� d���k (<3,0 g/dL) olanlarda MMAE klirensinin 2 kat daha d���k oldu�una i�aret etmi�tir.
Karaci�er yetmezli�i:
Bir �al��mada hafif (Child-Pugh A; n=1), orta (Child-Pugh B; n=5) ve �iddetli (Child-Pugh C; n=1) karaci�er yetmezli�i olan hastalara 1,2 mg/kg ADCETR�S uygulamas�ndan sonra ADCETR�S ve MMAE'nin farmakokineti�i de�erlendirilmi�tir. Hepatik fonksiyonu norma l olan hastalar ile kar��la�t�r�ld���nda karaci�er yetmezli�i olan hastalarda MMAE maruziyeti yakla��k 2,3 kat y�kselmi�tir (%90 GA 1,27-4,12 kat).
B�brek yetmezli�i
Bir �al��mada hafif (n=4), orta (n=3) ve �iddetli (n=3) b�brek yetmezli�i olan hastalara 1,2 mg/kg ADCETR�S uygulamas�ndan sonra ADCETR�S ve MMAE'nin farmakokineti�i de�erlendirilmi�tir. B�brek fonksiyonu normal olan hastalar ile kar��la�t�r�ld���nda �iddetli b�brek yetmezli�i (kreatinin klirensi <30 mL/dk) olan hastalarda MMAE maruziyeti yakla��k 1,9 kat y�kselmi�tir (%90 GA 0,85- 4,21 kat). Hafif veya orta dereceli b�brek yetmezli�i olan hastalarda herhangi bir etki g�zlenmemi�tir.
Geriyatrik pop�lasyon:
ADCETR�S'in pop�lasyon farmakokineti�i; 87 ya��na kadar 380 hastadan al�nan veriler dahil olmak �zere bir�ok �al��mada incelenmi�tir (≥65-<75 ya�lar�nda 34 hasta ve ≥75 ya�lar�nda 17 hasta). Ayr�ca, AVD ile kombinasyon halinde uygulanan brentuximab vedotinin pop�lasyon farmakokineti�i 82 ya��na kadar 661 hasta (≥65-<75 ya�lar�nda 42 hasta ve ≥75 ya�lar�nda 17 hasta) verisi �zerinden de�erlendirilmi�tir. Ya��n farmakokinetik �zerindeki etkisi her bir analizde incelenmi� ve �nemli bir e�de�i�ken fakt�r olmad��� bulunmu�tur.
Pediyatrik pop�lasyon:
Monoterapi
C25002
Faz 1/2 �al��mas�nda, ADCETR�S'in antikor ila� konjugat� (A�K) ve antimikrot�b�l ajan monometil auristatin E (MMAE) farmakokineti�i her 3 haftada bir uygulanan 30 dakikal�k 1,4 mg/kg veya 1,8 mg/kg ADCETR�S inf�zyonunu sonras�nda 36 pediyatrik (7-17 ya�lar�nda) n�kseden veya tedaviye diren�li HL ve sALCL hastas�nda (7-11 ya�lar�nda �ocuk n=12 ve 12-17 ya�lar�nda adolesan n=24) de�erlendirilmi�tir (bkz. b�l�m 5.1). A�K Ctipik olarak inf�zyonun sonunda veya inf�zyonun sonuna en yak�n numune alma zaman�nda g�zlenmi�tir. A�K'n�n serum konsantrasyonunda multi- eksponansiyonel d�����nde terminal yar�lanma �mr� yakla��k olarak 4 veya 5. g�nde g�zlenmi�tir. Maruziyetlerin, �al��ma pop�lasyonundaki d���k ya� /v�cut a��rl��� olan hastalar�n A�K maruziyetleriyle yakla��k olarak doz orant�l� oldu�u g�zlenmi�tir. Bu �al��madaki �ocuk ve ergenlerde medyan A�K EAA yeti�kinlere g�re yakla��k olarak s�ras�yla %14 ve %3 daha az olmu�tur, bununla birlikte MMAE maruziyetleri s�ras�yla yeti�kinlere g�re %53 daha az ve %13 daha fazla olmu�tur. Tek doz 1,8 mg/kg dozu uygulamas� sonras�nda antikor ila� konjugat� (A�K) i�in medyan C, EAA ve T12 ya��ndan k���k hastalarda s�ras�yla 29,8 µg/ml ve 67,9
µg*g�n/mL ve 12 ya� ve �zerinde olan hastalarda s�ras�yla 34,4 µg/mLve 77,8 µg*g�n/mL'dir. Tek doz 1,8 mg/kg dozu uygulamas� sonras�nda antimikrot�b�l ajan monometil auristatin E (MMAE) i�in medyan C, EAA ve T12 ya��ndan k���k hastalarda s�ras�yla 3,73 µg/ml ve 1 7,3 µg*g�n/mL ve 1,92 g�n olup 12 ya� ve �zerinde olan hastalarda s�ras�yla 6,33 µg/mLve 42,3 ng*g�n/mL ve1,82 g�n'd�r. A�A pozitif oldu�u etyit edilen pediyatrik hastalarda ADCETR�S'in klirensinde artma trendi g�r�lm��t�r. 12 ya��ndan k���k hastalar�n hi�birinde (11 hastada 0 hasta) ve 12 ya��nda ve daha b�y�k olan 2 hastada (23 hastada 2 hasta) anti ila� antikoru (A�A) kal�c� olarak pozitif hale gelmi�tir.
Kombinasyon tedavisi
C25004
Doksorubisin, vinblastin ve dakarbazin (AVD) ile kombinasyon halinde her 2 haftada bir 30 dakika boyunca 48 mg/m uygulanan BV'nin brentuximab vedotin ADC ve MMAE'nin farmakokineti�i, yeni te�his edilmi� ileri evre CD30+ klasik Hodgkin lenfomal� 6-17 ya� aras� 59 hastan�n (6-11 ya� aras� �ocuklar n=11 ve 12-17 ya� aras� adolesanlar n=48) yer ald��� faz 1/2 klinik �al��mada de�erlendirildi.
�nf�zyonun sonlar�na do�ru serumda ADC'nin C'� ger�ekle�ti ve yakla��k 4 g�nl�k yar�lanma �mr� ile multi-eksponansiyel bir �ekilde azald�. BV uygulamas�ndan yakla��k 2 g�n sonra plazmada MMAE'nin C'� ger�ekle�ti ve yar�lanma �mr� 2 g�ne yak�n oldu.
Tek bir 48 mg/m dozdan sonra ADC'nin geometrik ortalama C'� 22,5 µg/mL ve EEA's� 46,7 µg*g�n/mL idi. Tek bir 48 mg/m dozdan sonra MMAE'nin geometrik ortalama C'� 4,9 ng/mL ve EEA's� 27,2 ng*g�n/mL idi. Pediyatrik ya� gruplar�nda (< 12 ya�, 12 – 16 ya� ve > 16 ya�) AVD ile kombinasyon halinde 48 mg/m BV'nin v�cut alan�na g�re hesaplanan dozda verilmesinden sonra benzer ADC maruziyetleri elde edildi.
5.3. Klinik �ncesi g�venlilik verileri
Bir in vivo s��an kemik ili�i mikron�kleus �al��mas�nda MMAE'nin an�jenik �zellikleri oldu�u g�sterilmi�tir. Bu sonu�lar, h�crelerde MMAE'nin mitotik aparat �zerindeki farmakolojik etkisi (mikrot�b�l a��n� bozma) ile uyumlu olmu�tur.
ADCETR�S'in insanda erkek ve kad�n fertilitesi �zerindeki etkileri �al���lmam��t�r. Di�er yandan, s��andaki tekrarl� doz toksisitesi �al��malar�n�n sonu�lar� ADCETR�S i�in erkekte �reme fonksiyonlar�n� ve fertiliteyi olumsuz etkileme potansiyeline i�aret etmektedir. Testik�ler atrofi ve dejenerasyon, 16 haftal�k tedavisiz d�nem sonras�nda k�smen geri d�n��l� olmu�tur.
ADCETR�S, gebe di�i s��anlarda embriyo-fetal letaliteye neden olmu�tur.
Klinik d��� �al��malarda lenfoid deplesyonu ve timus a��rl���nda azalma g�zlenmi� olup bu bulgu, ADCETR�S kaynakl� MMAE'nin yol a�t���, mikrot�b�llerdeki farmakolojik bozulma ile uyumludur.
6. FARMAS�T�K �ZELL�KLER
6.1. Yard�mc� maddelerin listesi
Sitrik asit monohidrat (pH ayarlama ama�l�) Sodyum sitrat dihidrat (pH ayarlama ama�l�) α,α-Trehaloz dihidrat
Polisorbat 80
6.2. Ge�imsizlikler
Ge�imlilik �al��malar� bulunmad���ndan bu �r�n, b�l�m 6.6'da belirtilen d���nda di�er t�bbi �r�nler
ile kar��t�r�lmamal�d�r.
6.3. Raf �mr�
48 ay
Kullan�ma haz�rlama/suland�rma sonras�nda mikrobiyolojik a��dan �r�n hemen kullan�lmal�d�r. Bununla birlikte 2°C-8°C'de 24 saate kadar kimyasal ve fiziksel kullan�m i�i stabilite g�sterilmi�tir.
6.4. Saklamaya y�nelik �zel tedbirler
Saklamaya y�nelik �zel tedbirler tedbirler
Buzdolab�nda (2°C-8°C) saklay�n�z. Dondurmay�n�z.
I��ktan korumak i�in flakonu orijinal ambalaj�nda saklay�n�z.
T�bbi �r�n�n kullan�ma haz�rlanmas� ve seyreltilmesi sonras�ndaki saklama ko�ullar� i�in, bkz.
b�l�m 6.3.
6.5. Ambalaj�n niteli�i ve i�eri�i
B�til kau�uk t�pal� ve al�minyum/plastik ge�me contal�, 50 mg toz i�eren tip I cam flakon.
1 flakonluk ambalaj.
6.6. Be�eri t�bbi �r�nden arta kalan maddelerin imhas� ve di�er �zel �nlemler
Genel �nlemler
Anti-kanser ila�lar�n�n uygun saklanmas� ve imhas�na ili�kin prosed�rler g�z �n�nde bulundurulmal�d�r.
Bu t�bbi �r�n ile t�m i�lemler s�ras�nda uygun aseptik teknik uygulanmal�d�r. Kullan�ma haz�rlama talimatlar�
Her bir tek kullan�ml�k flakon 10,5 mL enjeksiyonluk su ile 5 mg/mL son konsantrasyona
seyreltilmelidir. Her bir flakonda, flakon ba��na 55 mg ADCETR�S ve toplam 11 mL suland�r�lm�� hacim sa�layan %10 fazlal�k bulunur.
Ak�� do�rudan keke veya toza de�il, flakonun �eperine y�nlendirilir. ��z�nmeye yard�mc� olmak i�in hafif�e kendi �evresinde d�nd�r�l�r. �ALKALANMAMALIDIR.
Flakondaki suland�r�lm�� ��zelti berrak ila hafif opak, renksiz ��zelti olup son pH de�eri 6,6'd�r. Suland�r�lm�� ��zelti yabanc� partik�l madde ve/veya renk bozuklu�u a��s�ndan g�zle incelenmelidir. Bunlardan herhangi biri g�zleniyorsa , t�bbi �r�n at�lmal�d�r.
�nf�zyon ��zeltisinin haz�rlanmas�
Uygun miktarda suland�r�lm�� ADCETR�S flakondan (flakonlardan) �ekilmeli ve 0,4-1,2 mg/mL ADCETR�S son konsantrasyonu elde etmek �zere sodyum klor�r 9 mg/mL (%0,9) enjeksiyonluk ��zelti i�eren bir inf�zyon torbas�na eklenmelidir. �nerilen seyrelti hacmi 150 mL'dir. �nceden suland�r�lm�� ADCETR�S ayr�ca %5 enjeksiyonluk dekstroz veya enjeksiyonluk laktatl� Ringer ile seyreltilebilir.
ADCETR�S i�eren ��zeltinin kar��t�r�lmas� i�in torba hafif�e ters �evrilir. �ALKALANMAMALIDIR.
Seyreltilecek hacim �ekildikten sonra flakonda kalm�� k�s�m varsa, yerel gerekliliklere g�re bertaraf edilmelidir.
Haz�rlanan ADCETR�S inf�zyon ��zeltisine veya intraven�z inf�zyon setine ba�ka bir t�bbi �r�n eklenmemelidir. Uygulama sonras�nda inf�zyon hatt� sodyum klor�r 9 mg/mL (%0,9) enjeksiyonluk ��zelti, %5 enjeksiyonluk dekstroz veya enjeksiyonluk laktatl� Ringer ile y�kanmal�d�r.
Seyreltildikten sonra ADCETR�S ��zeltisi derhal, �nerilen inf�zyon h�z�yla inf�ze edilir. Kullan�ma haz�rlama ile inf�zyon aras�nda ��zeltinin toplam saklama s�resi 24 saati ge�memelidir.
Doz miktar�n�n belirlenmesi:
Ek seyreltme yap�lacak toplam ADCETR�S dozunun (mL) belirlenmesi i�in hesaplama (bkz. b�l�m 4.2):
ADCETR�S dozu (mg/kg) x hastan�n beden a��rl��� (kg) | = Daha fazla seyreltilecek toplam ADCETR�S dozu (mL) |
Suland�r�lan flakon konsantrasyonu (5 mg/mL) |
Not: E�er hastan�n beden a��rl��� 100 kg'�n �zerinde ise doz hesaplamas�nda 100 kg kullan�lmal�d�r. Maksimum �nerilen doz 180 mg'd�r.
Gereken toplam ADCETR�S flakonu say�s�n�n belirlenmesi i�in hesaplama:
![]()
Uygulanacak toplam ADCETR�S dozu (mL)
Flakon ba��na toplam hacim (10 mL/flakon)
= Gereken ADCETR�S flakon say�s�
Tablo 18: 60 kg-120 kg aral���ndaki beden a��rl�klar� i�in �nerilen 1,8 mg/kg, 1,2 mg/kg veya 0,9 mg/kg ADCETR�S dozunu alan hastalarda �rnek hesaplamalar
Tavsiye edilen doz | Hasta a��rl��� (kg) | Toplam doz = hasta a��rl���n�n �nerilen doz ile �arp�m� | Seyreltilecek toplam hacim = toplam dozun rekonstit�e flakon konsantrasyonuna [5 mg/mL] b�l�m� | Gereken flakon say�s� = seyreltilecek toplam hacmin, her bir flakon toplam hacmine [10 mL/flakon] b�l�m� |
1,8 mg/kg (maksimum 180 mg'a kadar) | 60 kg | 108 mg | 21.6 mL | 2.16 flakon |
80 kg | 144 mg | 28.8 mL | 2.88 flakon | |
100 kg | 180 mg | 36 mL | 3.6 flakon | |
120 kg | 180 mg | 36 mL | 3.6 flakon | |
1,2 mg/kg (maksimum 120 mg'a kadar) | 60 kg | 72 mg | 14.4 mL | 1.44 flakon |
80 kg | 96 mg | 19.2 mL | 1.92 flakon | |
100 kg | 120 mg | 24 mL | 2.4 flakon | |
120 kg | 120 mg | 24 mL | 2.4 flakon | |
0,9 mg/kg (maksimum 90 mg'a kadar) | 60 kg | 54 mg | 10.8 mL | 1.08 flakon |
80 kg | 72 mg | 14.4 mL | 1.44 flakon | |
100 kg | 90 mg | 18 mL | 1.8 flakon | |
120 kg | 90 mg | 18 mL | 1.8 flakon |
 Travma Sonras� Bunal�m�
Travmatik bir olay, g�nl�k ola�an olaylar�n d���nda olan ve ki�iyi derinden
rahats�z eden bir olayd�r.Bir�ok olay b�yle bir etki g�sterebilir.
Travma Sonras� Bunal�m�
Travmatik bir olay, g�nl�k ola�an olaylar�n d���nda olan ve ki�iyi derinden
rahats�z eden bir olayd�r.Bir�ok olay b�yle bir etki g�sterebilir. |
 �nme
�nme, beynin hasar g�rmesinin sonucudur. Bu hasar, beynin bir k�sm�ndaki ya bir kanama
ya da akut kan eksikli�i nedeniyle o k�sm�n ge�ici ya da kal�c� olarak i�levini yapamamas�na
yol a�ar.
�nme
�nme, beynin hasar g�rmesinin sonucudur. Bu hasar, beynin bir k�sm�ndaki ya bir kanama
ya da akut kan eksikli�i nedeniyle o k�sm�n ge�ici ya da kal�c� olarak i�levini yapamamas�na
yol a�ar. |
 |
En Yayg�n Alerji T�rleri Ba����kl�k sistemi, polen, ar� zehiri veya evcil hayvan gibi yabanc� bir maddeye veya �o�u insanda reaksiyona neden olmayan bir yiyece�e tepki g�sterdi�inde alerjiler meydana gelir. |
 |
Omurilik zedelenmeleri Omurilik zedelenmesini takip eden birka� g�n i�inde, hi�kimse hasarin ne kadar olacagini tahmin edemez. Buradaki sorun, omuriligin herhangi bir zedelenmesinden hemen sonra, bir omurilik sokunun olusmasidir. |
 |
Belso�uklu�u, Chlamydia ve Frengi Belso�uklu�u, bakterilerin sebep oldu�u bir enfeksiyondur. Cinsel ili�ki yoluyla bula��r ve d�lyata�� boynunda, idrar yollar�nda, an�ste, makatta ve bo�azda enfeksyona sebep olabilir. |
�LA� GENEL B�LG�LER�
Takeda �la�lar� ve Ticaret Ltd.�ti.
| Geri �deme Kodu | A16640 |
| Sat�� Fiyat� | 93066.89 TL [ 3 Aug 2026 ] |
| �nceki Sat�� Fiyat� | 93066.89 TL [ 27 Jul 2026 ] |
| Original / Jenerik | Original �la� |
| Re�ete Durumu | Normal Re�eteli bir ila�d�r. |
| Barkodu | 8699456790058 |
| Etkin Madde | Brentuksimab Vedotin |
| ATC Kodu | L01XC12 |
| Birim Miktar | 50 |
| Birim Cinsi | MG |
| Ambalaj Miktar� | 1 |
| Antineoplastik ve �mm�nomod�lat�r Ajanlar > Di�er Kanser �la�lar� |
| �thal ( ref. �lke : Almanya ) ve Be�eri bir ila�d�r. |