XGEVA 120 mg SC. enjeksiyonluk çöz. içeren 1 flakon Kısa Ürün Bilgisi
{ Donesumab }
1. BEŞERİ TIBBİ ÜRÜNÜN ADI
XGEVA® 120 mg SC enjeksiyonluk çözelti Steril
2. KALİTATİF VE KANTİTATİF BİLEŞİM
Etkin madde
Her flakon, 1,7 ml çözeltide 120 mg denosumab içerir (70 mg/ml).
Denosumab, rekombinant DNA teknolojisi kullanılarak bir memeli hücre dizisinde (CHO) üretilen bir insan monoklonal IgG2 antikorudur.
Yardımcı maddeler
Her 1,7 ml çözelti, 78 mg sorbitol (E420) ve sodyum (pH ayarı için sodyum hidroksit olarak) içerir.
Yardımcı maddelerin tam listesi için, bölüm 6.1'e bakınız.
3. FARMASÖTİK FORMU
Enjeksiyonluk çözelti.
Renksiz ile hafif sarı arası renkte, berrak bir çözeltidir ve eser miktarda şeffaf ile beyaz arası renkte proteinli parçacıklar içerebilir.
4. KLİNİK ÖZELLİKLER
4.1. Terapötik endikasyonlar
Daha önce zoledronik asit kullanmamış, kırık veya bası riski yüksek vertebra, femur, humerus gibi yük taşıyan kemiklere metastaz yapmış meme kanseri, hormon refrakter prostat kanseri veya küçük hücreli dışı akciğer kanseri hastalarında kemik progresyonuna kadar kullanımı endikedir. Kemik progresyonu sonrası veya iskeletle ilişkili olay gelişimi sonrası denosumab tedavisine devam edilemez.
Kemikte dev hücreli tümörün cerrahi yöntemlerle çıkartılamadığı veya cerrahi uygulamanın şiddetli morbidite ile sonuçlanmasının muhtemel olduğu yetişkinlerin ve iskeleti olgunlaşmış adolesanların tedavisinde endikedir.
Multipl miyelom tanısı konmuş olup antimiyelom tedavi endikasyonu olan, kreatinin klerensi 30 ml/dk altında olan hastalarda miyelom ilişkili kemik komplikasyonlarının önlenmesinde endikedir.
4.2. Pozoloji ve uygulama şekli
XGEVA®, bir sağlık mesleği mensubunun sorumluluğu altında uygulanmalıdır.
Pozoloji/uygulama sıklığı ve süresi:
Hiperkalsemi bulunmaması şartıyla, tüm hastalara günlük en az 500 mg kalsiyum ve 400 IU D vitamini takviyesi gereklidir (bkz. bölüm 4.4).
İleri evre malignitesi bulunan ve kemik tutulumu olan yetişkinlerde iskeletle ilişkili olayların önlenmesi için
Önerilen doz, 4 haftada bir, tek doz halinde uyluğa, karın duvarına ya da üst kola uygulanan
120 mg'lık subkütan enjeksiyondur.
Dev hücreli kemik tümörü
XGEVA® için önerilen doz, 4 haftada bir, tek doz halinde uyluğa, karna ya da üst kola
uygulanan 120 mg'lık subkütan enjeksiyon ve ilave olarak tedavinin 8. gününde ve
15. gününde 120 mg'lık dozlardır.
Faz II çalışmada olan, dev hücreli kemik tümörünün tamamen rezeksiyonu uygulanan hastalar, çalışma protokolü uyarınca, ameliyattan sonra 6 ay daha ilave tedavi almışlardır.
Dev hücreli kemik tümörü bulunan hastalar, tedaviden fayda görmeye devam edip etmediklerinin belirlenmesi için düzenli aralıklarla değerlendirilmelidir. Hastalığı XGEVA® ile kontrol altına alınan hastalarda, tedaviye ara verilmesi veya tedavinin kesilmesi değerlendirilmemiştir; ancak, bu hastalardaki sınırlı veriler, tedavinin kesilmesiyle yoksunluk etkisi olduğuna işaret etmemektedir.
Multipl miyelom
1 yılın sonunda endikasyonu tekrar değerlendirilerek remisyonda olmayan, miyelom tedavisi devam eden hastalarda en fazla 2 yıl kullanılır.
Uygulama şekli:
Subkütan kullanıma yöneliktir.
Kullanım, taşıma ve imha talimatları bölüm 6.6'da verilmiştir.
Özel popülasyonlara ilişkin ek bilgiler:
Böbrek yetmezliği:
Böbrek yetmezliği olan hastalarda doz ayarlaması gerekli değildir (kalsiyum izlenmesi ile ilgili öneriler için bkz. bölüm 4.4, 4.8 ve 5.2).
Karaciğer yetmezliği:
Denosumabın karaciğer yetmezliği bulunan hastalardaki güvenliliği ve etkililiği incelenmemiştir (bkz. bölüm 5.2).
Pediyatrik popülasyon:
XGEVA®'nın güvenliliği ve etkililiği, iskeleti olgunlaşmış, dev hücreli kemik tümörü bulunan ergenler (12-17 yaş grubu) hariç, pediyatrik hastalarda (< 18 yaş) ortaya konmamıştır.
XGEVA®, iskeleti olgunlaşmış, dev hücreli kemik tümörü bulunan ergenler (12-17 yaş grubu) hariç, pediyatrik hastalarda (< 18 yaş) önerilmez (bkz. bölüm 4.4).
Rezeke edilemeyen veya cerrahi rezeksiyonun şiddetli morbidite ile sonuçlanması yüksek olasılıklı olan, dev hücreli kemik tümörü bulunan iskeleti olgunlaşmış ergenlerin tedavisinde pozoloji yetişkinlerdekiyle aynıdır.
Hayvan çalışmalarında RANK/RANK ligandının (RANKL) inhibisyonu, kemik büyümesinin inhibisyonuyla ve diş sürmesinde eksiklikle eşleştirilmiş ve bu değişimlerin RANKL inhibisyonunun kesilmesiyle kısmen geri döndürülebilir olduğu görülmüştür (bkz. bölüm 5.3).
Geriyatrik popülasyon (≥ 65 yaş):
Yaşlı hastalarda doz ayarlaması gerekmemektedir (bkz. bölüm 5.2).
4.3. Kontrendikasyonlar
Etkin madde
Her flakon, 1,7 ml çözeltide 120 mg denosumab içerir (70 mg/ml).
Şiddetli, tedavi edilmemiş hipokalsemi (bkz. bölüm 4.4).
Dental cerrahi veya ağız cerrahisi kaynaklı iyileşmemiş lezyonlar.
4.4. Özel kullanım uyarıları ve önlemleri
Kalsiyum ve D Vitamini Takviyesi
Hiperkalsemi bulunmaması şartıyla, tüm hastalara kalsiyum ve D vitamini takviyesi gereklidir (bkz. bölüm 4.2).
Hipokalsemi
XGEVA® tedavisine başlamadan önce, önceden var olan hipokalsemi tedavi edilmelidir. Hipokalsemi, XGEVA® tedavisi sırasında herhangi bir zamanda meydana gelebilir. Aşağıdaki hallerde kalsiyum seviyelerinin izlenmesi gerekmektedir;
XGEVA®'nın ilk dozu verilmeden önce,
4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleri
Etkileşim çalışmaları yapılmamıştır.
XGEVA®, klinik araştırmalarda standart anti-kanser tedavileriyle kombine halde ve daha önce bisfosfonat alan gönüllülerde kullanılmıştır. Eşzamanlı kemoterapi ve/veya hormon tedavisi ya da önceden yaşanan intravenöz bisfosfonat maruziyeti nedeniyle denosumabın dip serum konsantrasyonunda ve farmakodinamiğinde (kreatinin ayarlamalı üriner N-telopeptid, uNTx/Cr) klinik açıdan anlamlı bir değişim olmamıştır.
Özel popülasyonlara ilişkin ek bilgiler
Etkileşim çalışmaları yapılmamıştır.
Pediyatrik popülasyon:
Etkileşim çalışmaları yapılmamıştır.
4.6. Gebelik ve laktasyon
Gebelik kategorisi: C
Çocuk doğurma potansiyeli bulunan kadınlar/Doğum kontrolü (Kontrasepsiyon) XGEVA®'nın hamile kalma potansiyeline sahip, doğum kontrolü kullanmayan kadınlarda kullanımı tavsiye edilmemelidir.
Gebelik dönemi
XGEVA®'nın gebe kadınlarda kullanımına ilişkin yeterli veri mevcut değildir. Hayvanlar üzerinde yapılan araştırmalar üreme toksisitesi göstermiştir. İnsanlar üzerindeki potansiyel risk bilinmemektedir.
XGEVA®'nın hamile kadınlarda kullanılması tavsiye edilmemektedir.
Kadınlar, XGEVA® tedavisi sırasında ve XGEVA® tedavisinden sonra en az 5 ay süreyle hamile kalmamalıdır. Monoklonal antikorlar, hamilelik ilerledikçe lineer olarak ve en fazla üçüncü trimesterde plesentaya taşındığından, XGEVA®'nın etkisinin hamileliğin ikinci ve üçüncü trimesterinde daha çok görülmesi muhtemeldir.
Laktasyon dönemi
Denosumab'ın insan sütüyle atılıp atılmadığı bilinmemektedir. Yenidoğanlar/bebekler için risk göz ardı edilemez. Knockout fare çalışmaları, gebelik sırasında RANKL eksikliğinin meme bezlerinin gelişimini engelleyebileceğini ve bunun da post-partum süreçte laktasyon yetmezliğine yol açabileceğini göstermektedir (bkz. bölüm 5.3).
Emzirmenin ya da XGEVA® tedavisinin durdurulup durdurulmayacağına/tedaviden kaçınılıp kaçınılmayacağına ilişkin karar verilirken, emzirmenin çocuk açısından faydası ve XGEVA® tedavisinin emziren anne açısından faydası dikkate alınmalıdır.
Üreme yeteneği/Fertilite
Denosumabın insan fertilitesine etkileri hakkında veri mevcut değildir. Hayvan çalışmaları, fertiliteyle ilgili doğrudan ya da dolaylı zararlı etkileri hakkında bir kanıt sunmamaktadır (bkz. bölüm 5.3).
4.7. Araç ve makine kullanımı üzerindeki etkiler
XGEVA®'nın araç ve makine kullanımı üzerine etkisi yoktur veya ihmal edilebilir düzeydedir.
4.8. İstenmeyen etkiler
Güvenlilik profilinin özeti
Genel güvenlilik profili, XGEVA®'nın onaylı tüm endikasyonlarında tutarlıdır.
XGEVA® uygulaması sonrasında, çoğunlukla ilk 2 hafta içinde olmak üzere, hipokalsemi geliştiği çok sık olarak bildirilmiştir. Hipokalsemi şiddetli ve semptomatik olabilir (bkz. bölüm 4.8 - Seçilmiş advers etkilerin açıklamaları). Serum kalsiyumundaki düşüşler genellikle kalsiyum ve D vitamini takviyesiyle uygun şekilde yönetilmiştir. XGEVA® ile görülen en yaygın advers reaksiyonlar kas-iskelet ağrısıdır. Çene osteonekrozu olguları (bkz. bölüm 4.4 ve bölüm 4.8 - Seçilmiş advers etkilerin açıklamaları) XGEVA® kullanan hastalarda yaygın şekilde gözlenmiştir.
Advers etkilerin tablo halinde listesi
Dört faz III ve iki faz II klinik çalışmadaki ve pazarlama sonrası deneyim insidans oranlarına dayanarak advers etkilerin sınıflandırılması için aşağıdaki yöntem kullanılmıştır (bkz. Tablo 1): çok yaygın (≥ 1/10), yaygın (≥ 1/100 ila < 1/10 arası), yaygın olmayan (≥ 1/1.000 ila < 1/100), seyrek (≥ 1/10.000 ila < 1/1.000), çok seyrek (< 1/10.000) ve bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor). Advers etkiler, her sıklık grubu ve sistem organ sınıfı için azalan ciddiyet sırasına göre sunulmuştur.
Tablo 1. Kemik tutulumlu ilerlemiş malignitelere sahip olan, multipl miyelomu veya dev hücreli kemik tümörü bulunan hastalarda bildirilen advers etkiler
MedDRA sistem organ sınıfı | Sıklık kategorisi | Advers etkiler |
(Kist ve polipler de dahil olmak uzere) iyi huylu ve kötü huylu neoplazmalar | Yaygın | Yeni primer malignite |
Bağışıklık sistemi hastalıkları | Seyrek | İlaç hipersensitivitesi |
Seyrek | Anafilaktik reaksiyon | |
Metabolizma ve beslenme hastalıkları | Çok yaygın | Hipokalsemi |
Yaygın | Hipofosfatemi | |
Yaygın olmayan | Dev hücreli kemik tümörü bulunan hastalarda tedavinin bırakılmasının ardından hiperkalsemi | |
Solunum, göğüs bozuklukları ve mediastinal hastalıkları | Çok yaygın | Dispne |
Gastrointestinal hastalıkları | Çok yaygın | Diyare |
Yaygın | Diş çekimi |
MedDRA sistem organ sınıfı | Sıklık kategorisi | Advers etkiler |
Deri ve deri altı dokusu hastalıkları | Yaygın | Hiperhidroz |
Yaygın | Alopesi | |
Yaygın olmayan | Likenoid ilaç erüpsiyonları | |
Kas-iskelet bozuklukları, bağ dokusu ve kemik hastalıkları | Çok yaygın | Kas-iskelet ağrısı |
Yaygın | Çene osteonekrozu | |
Yaygın olmayan | Atipik femur kırıkları | |
Bilinmiyor | Dış kulak yolunda osteonekroz |
Seçilmiş advers etkilerin açıklamaları
Hipokalsemi
İskeletle ilişkili olayları (SRE) önleme klinik çalışmalarında denosumab tedavisi uygulanan gönüllülerde hipokalsemi insidansı zoledronik asit alan gönüllülere kıyasla daha yüksek bulunmuştur.
En yüksek hipokalsemi insidansı multipl miyelom hastalarıyla yapılan faz III çalışmada gözlemlenmiştir. Hipokalsemi, XGEVA® tedavisi alan hastaların %16,9'unda, zoledronik asit tedavisi alan hastaların %12,4'ünde bildirilmiştir. XGEVA® tedavisi alan hastaların
%1,4'ünde ve zoledronik asit tedavisi alan hastaların %0,6'sında serum kalsiyum düzeyinde
3. derece azalma saptanmıştır. Serum kalsiyum düzeyinde 4. derece azalma ise XGEVA® tedavisi alan hastaların %0,4'ünde ve zoledronik asit tedavisi alan hastaların %0,1'inde tespit edilmiştir.
Kemik tutulumlu ilerlemiş malignitesi bulunan hastalarla yapılan aktif-kontrollü üç adet faz III klinik araştırmada, XGEVA® ile tedavi edilen hastaların %9,6'sında, zoledronik asit ile tedavi edilen hastaların ise %5'inde hipokalsemi bildirilmiştir.
XGEVA® ile tedavi edilen hastaların %2,5'inde, zoledronik asit ile tedavi edilen hastaların ise %1,2'sinde serum kalsiyum seviyelerinde 3. derece düşüş görülmüştür. XGEVA® ile tedavi edilen hastaların %0,6'sında, zoledronik asit ile tedavi edilen hastaların ise %0,2'sinde serum kalsiyum seviyelerinde 4. derece düşüş görülmüştür (bkz. bölüm 4.4).
Dev hücreli kemik tümörü bulunan hastalarda gerçekleştirilen iki adet faz II tek kollu klinik çalışmada, hastaların %5,7'sinde hipokalsemi bildirilmiştir. Bu advers olayların hiçbiri ciddi kabul edilmemiştir.
Pazarlama sonrası deneyimde, ciddi semptomatik hipokalsemi vakaları (ölümcül vakalar dahil) bildirilmiş olup olguların büyük bir kısmı tedavinin ilk haftalarında meydana gelmiştir. Örnek olarak şiddetli semptomatik hipokalseminin klinik bulguları arasında QT aralığında uzama, tetani, nöbetler ve mental durum değişikliği (koma dahil) yer almıştır (bkz. bölüm 4.4). Klinik çalışmalardaki hipokalsemi semptomları ise parastezi veya kaslarda sertlik, seğirme, spazmlar ve kas kramplarını içermektedir.
Çene osteonekrozu (ÇO)
Klinik çalışmalarda, ÇO insidansının daha uzun süreli maruziyetle birlikte arttığı görülmüştür; ayrıca XGEVA® tedavisi durdurulduktan sonra da ÇO tanısı konulanlar olmuş, olguların büyük bölümü son dozdan sonraki 5 ay içerisinde ortaya çıkmıştır. ÇO veya çene osteomyeliti öyküsü olan, ağız cerrahisi gerektiren aktif dental veya çene rahatsızlığı bulunan, iyileşmemiş dental/ağız cerrahisi olan veya herhangi bir invazif diş prosedürü planlanmış olan hastalar klinik araştırmalara dahil edilmemiştir.
İskeletle ilişkili olayları (SRE) önleme klinik çalışmalarında denosumab tedavisi uygulanan gönüllülerde ÇO insidansı zoledronik asit alan gönüllülere kıyasla daha yüksek bulunmuştur. En yüksek ÇO insidansı multipl miyelom hastalarıyla yapılan faz III çalışmada gözlemlenmiştir. Bu çalışmanın çift kör tedavi fazında XGEVA® tedavisi alan (medyan maruziyet 19,4 ay; aralık 1-52) hastaların %5,9'unda ve zoledronik asit tedavisi alan hastaların %3,2'sinde ÇO doğrulanmıştır. Bu çalışmanın çift kör tedavi fazı tamamlandığında, XGEVA® grubunda (medyan maruziyet 19,4 ay; aralık 1-52) doğrulanmış ÇO'nun hasta yılına göre ayarlanmış insidansı 100 hasta yılı için tedavinin ilk yılında 2, ikinci yılında 5 ve sonrasında 4,5 olarak belirlenmiştir. ÇO'ye kadar geçen medyan süre 18,7 ay (aralık: 1-44) olarak kaydedilmiştir.
Kemik tutulumlu ilerlemiş malignitesi bulunan hastalarla yapılan aktif-kontrollü üç adet faz III klinik araştırmanın primer tedavi aşamalarında, XGEVA® (medyan maruziyet 12 ay; aralık: 0,1-40,5) ile tedavi edilen hastaların %1,8'inde, zoledronik asit ile tedavi edilen hastaların ise %1,3'ünde ÇO görüldüğü bildirilmiştir. Bu vakaların klinik özellikleri, tedavi grupları arasında benzerlik göstermiştir. Doğrulanmış ÇO görülen gönüllülerin çoğunun (her iki tedavi grubunda %81), diş çekimi, zayıf ağız hijyeni ve/veya dental cihaz kullanımı öyküsü vardır. Gönüllülerin çoğu kemoterapi almaktadır ya da geçmişte almıştır.
Meme veya prostat kanserli hastalar üzerinde yapılan araştırmalara XGEVA® uzatma tedavisi fazı dahil edilmiştir (medyan genel maruziyet 14,9 ay; aralık: 0,1-67,2). ÇO, uzatma tedavisi fazı sırasında meme ve prostat kanserli hastaların %6,9'unda doğrulanmıştır.
Doğrulanan ÇO'nun hasta-yılına uyarlanmış genel insidansı 100 hasta yılı için tedavinin ilk yılında 1,1, ikinci yılında 3,7 ve sonrasında 4,6'dır. ÇO'ya kadar geçen ortalama süre 20,6 aydır (aralık: 4 - 53).
İsveç, Danimarka ve Norveç'te XGEVA® veya zoledronik asit ile tedavi edilen kanserli 2.877 hastada yapılan randomize olmayan, retrospektif, gözlemsel bir çalışma; tıbbi olarak doğrulanmış ÇO'nun 5 yıllık insidans oranlarının XGEVA® alan hastalardan oluşan bir kohortta %5,7 (%95 CI: 4,4, 7,3; 20 aylık medyan takip süresi [aralık 0,2-60]) ve zoledronik
asit alan hastalardan oluşan ayrı bir kohortta %1,4 (%95 CI: 0,8, 2,3; 13 aylık medyan takip süresi [aralık 0,1-60]) olduğunu göstermiştir. Zoledronik asitten XGEVA®'ya geçen hastalarda ÇO'nun 5 yıllık insidans oranı %6,6 (%95 CI: 4,2, 10; 13 aylık medyan takip süresi [aralık 0,2-60]) olmuştur.
Daha uzun tedavi maruziyeti (7 yıla kadar) ile metastatik olmayan prostat kanserli hastalarda (XGEVA®'nın endike olmadığı bir hasta popülasyonu) yapılan bir faz III çalışmasında hasta yılının ayarlandığı doğrulanmış ÇO insidansı; 100 hasta yılı için tedavinin birinci yılında 1,1, ikinci yılında 3 ve sonrasında 7,1 olmuştur.
Dev hücreli kemik tümörü bulunan hastaların yer aldığı uzun süreli faz II açık etiketli klinik çalışmada (Çalışma 6, bkz. bölüm 5.1), bir ergenin dahil olduğu hastaların %6,8'inde ÇO varlığı doğrulanmıştır (medyan doz sayısı: 34; aralık: 4 – 116). Çalışma tamamlandığında, güvenlilik takibi dahil olmak üzere çalışmada geçen medyan sürenin 60,9 ay olduğu belirlenmiştir (aralık: 0 – 112,6). Doğrulanmış ÇO'nun hasta yılına göre ayarlanmış insidansı 100 hasta yılı için toplam 1,5 olarak kaydedilmiştir (tedavinin ilk yılında 100 hasta yılı için 0,2, ikinci yılında 1,5, üçüncü yılında 1,8, dördüncü yılında 2,1, beşinci yılında 1,4 ve sonrasında 2,2). ÇO'ya kadar geçen medyan süre 41 ay olmuştur (aralık: 11 – 96)
İlaca bağlı aşırı duyarlılık reaksiyonları
Pazarlama sonrası deneyimde, XGEVA® tedavisi alan hastalarda, seyrek anafilaktik reaksiyonlar dahil olmak üzere, aşırı duyarlılık olayları bildirilmiştir.
Atipik femur kırıkları
Klinik araştırma programında, XGEVA® tedavisi alan hastalarda atipik femur kırıkları yaygın olmayan sıklıkta bildirilmiştir ve tedavinin uzun sürmesiyle bu risk artar. Atipik femur kırıkları, tedavi boyunca ve tedavinin kesilmesinden 9 ay sonrasına kadar görülmüştür (bkz. bölüm 4.4).
Likenoid ilaç erüpsiyonları
Pazarlama sonrası deneyimde hastalarda likenoid ilaç erüpsiyonları (örneğin, liken planus benzeri reaksiyonlar) bildirilmiştir.
Kas-iskelet ağrısı
Pazarlama sonrası deneyimde XGEVA® alan hastalarda şiddetli olgular dahil kas-iskelet ağrısı bildirilmiştir. Klinik çalışmalarda kas-iskelet ağrısı, hem denosumab hem de zoledronik asit tedavi gruplarında çok yaygın görülmüştür. Çalışma tedavisinin bırakılmasına yol açan kas-iskelet ağrısının yaygın olmadığı görülmüştür.
Yeni primer maligniteler
İleri evre malignitesi bulunan ve kemik tutulumu olan hastaların yer aldığı aktif kontrollü dört faz III klinik çalışmanın primer çift kör tedavi fazlarında, XGEVA® tedavisi alan (medyan maruziyet 13,8 ay; aralık. 1-51,7) 3.691 hastanın 54'ünde (%1,5) ve zoledronik asit
tedavisi alan (medyan maruziyet 12,9 ay; aralık: 1-50,8) 3.688 hastanın 33'ünde (%0,9) yeni primer malignite bildirilmiştir.
Birinci yılda kümülatif insidans sırasıyla denosumab için %1,1, zoledronik asit için %0,6 olmuştur.
Belirli bir kanser veya kanser grupları için tedaviyle ilgili herhangi bir model belirgin olmamıştır.
Pediyatrik popülasyon
XGEVA®, dev hücreli kemik tümörü bulunan, iskeleti olgunlaşmış 28 ergenin kaydedildiği açık etiketli bir çalışmada incelenmiştir. Bu sınırlı verilere dayanarak, advers olay profilinin yetişkinlere benzer olduğu görülmüştür.
Pediyatrik hastalarda, pazarlama sonrası deneyimde tedavinin bırakılmasının ardından klinik olarak anlamlı hiperkalsemi bildirilmiştir (bkz. bölüm 4.4).
Özel popülasyonlara ilişkin ek bilgiler:
Böbrek yetmezliği:
İlerlemiş kanseri bulunmayan, ciddi böbrek yetmezliği (kreatinin klerensi < 30 ml/dakika) bulunan ya da diyaliz alan hastalarla yapılan bir klinik çalışmada, kalsiyum takviyesi olmaması durumunda hipokalsemi gelişimi riskinin daha fazla olduğu görülmüştür. XGEVA® tedavisi sırasında hipokalsemi gelişme riski, böbrek yetmezliğinin derecesi arttıkça yükselmektedir. İleri derecede kanser hastası olmayan hastalarda yapılan bir klinik çalışmada ciddi böbrek yetmezliği olan (kreatinin klerensi < 30 ml/dakika) hastaların %19'unda ve diyaliz alan hastaların %63'ünde kalsiyum takviyesi almalarına rağmen hipokalsemi gelişmiştir. Klinik olarak anlamlı hipokalseminin genel insidansı %9 olmuştur.
Ayrıca ciddi böbrek yetmezliği bulunan veya diyaliz alan XGEVA® kullanan hastalarda paratiroid hormonda eşlik eden yükselmeler gözlenmiştir. Böbrek yetmezliği olan hastalarda kalsiyum düzeylerinin izlenmesi ile yeterli kalsiyum ve D vitamini alımı özellikle önemlidir (bkz bölüm 4.4).
Şüpheli advers reaksiyonların raporlanması
Ruhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar/risk dengesinin sürekli olarak izlenmesine olanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli advers reaksiyonu Türkiye Farmakovijilans Merkezi (TÜFAM)'ne bildirmeleri gerekmektedir. (www.titck.gov.tr; e-posta: tufam@titck.gov.tr; tel: 0 800 314 00 08; faks: 0 312 218 35 99).
4.9. Doz aşımı ve tedavisi
Klinik çalışmalarda doz aşımıyla ilgili bir deneyim bulunmamaktadır. XGEVA®, klinik çalışmalarda 4 haftada bir 180 mg'a kadar ve 3 haftada bir 120 mg'a kadar dozlarda uygulanmıştır.
5. FARMAKOLOJİK ÖZELLİKLER
5.1. Farmakodinamik özellikler
Farmakoterapötik grup: Kemik hastalıklarının tedavisinde kullanılan ilaçlar - Kemik yapısını ve mineralizasyonu etkileyen diğer ilaçlar.
ATC kodu: M05BX04.
Etki mekanizması
RANKL, transmembran ya da çözünür protein halinde bulunur. RANKL, resorpsiyondan sorumlu tek hücre tipi olan osteoklastların oluşumu, işlevi ve sağkalımı için önemlidir. RANKL tarafından uyarılan yüksek osteoklast aktivitesi, metastatik kemik hastalığında ve multipl miyelomda kemik yıkımı konusunda kilit öneme sahip bir mediyatördür. Denosumab, yüksek bir afiniteyle ve spesifik olarak RANKL'yi hedefleyen ve ona bağlanarak RANKL/RANK etkileşimini engelleyen ve osteoklast sayısının ve işlevinin azalmasına yol açan, böylece kemik resorpsiyonunu ve kanserden kaynaklanan kemik yıkımını azaltan bir insan monoklonal antikorudur (IgG2).
Dev hücreli kemik tümörleri, RANK ligandını eksprese eden neoplastik stromal hücreler ve RANK eksprese eden osteoklast benzeri dev hücrelerle karakterizedir. Dev hücreli kemik
tümörü bulunan hastalarda, denosumab RANK ligandına bağlanır ve osteoklast benzeri dev hücreleri anlamlı ölçüde azaltır veya ortadan kaldırır. Sonuç olarak, osteoliz azalır ve proliferatif tümör stromasının yerine non-proliferatif, farklılaşmış (diferansiye), yoğun dokulu yeni kemik gelir.
Farmakodinamik etkiler
Kemik tutulumu olan ileri evre malignitelerin bulunduğu hastalarla ilgili faz II klinik çalışmalarda, 4 haftada bir (Q4W) veya 12 haftada bir subkütan (SK) dozlamayla XGEVA® uygulanması kemik rezorpsiyonu (uNTx/Cr, serum CTx) belirteçlerinde hızlı bir azalmayla sonuçlanmış, önceden bifosfonat tedavisine veya başlangıçtaki uNTx/Cr düzeyine bağlı olmaksızın 1 hafta içinde uNTx/Cr için medyan azalmaların %80 oranında olduğu kaydedilmiştir. Kemik tutulumu olan ileri evre malignitelerin bulunduğu hastalarla ilgili faz III klinik çalışmalarda, yaklaşık %80 medyan uNTx/Cr azalmaları 49 haftalık XGEVA® (Q4W 120 mg) tedavisi boyunca kalıcı olmuştur.
İmmünojenisite
Klinik çalışmalarda, ilerlemiş kanseri olan veya dev hücreli kemik tümörü bulunan hastalarda XGEVA® için nötralize edici antikorlar gözlemlenmemiştir. Hassas bir immünolojik miktar tayini kullanılarak yapılan testlerde, 3 yıla kadar sürelerle denosumab tedavisi verilen hastaların %1'inden azı nötralize edici olmayan bağlanıcı antikorlar bakımından pozitif bulunmuş ve bunlarda farmakokinetik, toksisite ya da klinik yanıt bakımından bir farklılık kanıtına rastlanmamıştır.
Solid tümörlerden metastaz oluşmuş hastalarda klinik etkililiği ve güvenliliği
4 haftada bir uygulanan 120 mg XGEVA® SC ve 4 haftada bir uygulanan 4 mg IV zoledronik asidin (düşük renal fonksiyon için doz ayarlaması yapılmış) etkililiği ve güvenliliği, kemik tutulumlu ilerlemiş malignitesi bulunan IV-bisfosfonat kullanmamış hastalarla yapılan şu üç randomize, çift kör, aktif-kontrollü çalışmayla kıyaslanmıştır: Meme kanseri bulunan (çalışma 1), diğer solid tümörler ya da multipl miyelomu bulunan (çalışma 2) ve kastrasyona dirençli prostat kanseri bulunan erişkinler (çalışma 3). Bu aktif-kontrollü klinik çalışmalarda, 5.931 hastada güvenlilik değerlendirilmiştir. ÇO ya da çene osteomiyeliti öyküsü, ağız cerrahisi gerektiren aktif bir diş ya da çene sorunu, iyileşmemiş diş/ağız cerrahisi bulunan ya da invazif bir dental prosedür geçirmesi planlanan hastalar, bu çalışmalara dahil edilmemiştir. Primer ve sekonder sonlanım noktalarında, bir ya da daha fazla iskeletle ilişkili olayın (SRE) varlığı değerlendirilmiştir. XGEVA®'nın zoledronik aside üstünlüğünün kanıtlandığı çalışmalarda, hastalara önceden belirlenmiş 2 yıllık uzatma tedavisi fazında açık etiketli XGEVA® sunulmuştur. SRE aşağıdakilerden herhangi biri olarak tanımlanmıştır: patolojik kırık (vertebral veya vertebra dışı), kemiğe yönelik radyasyon tedavisi (radyoizotop kullanımı dahil), kemik ameliyatı veya omurilik sıkışmasından herhangi biri olarak SRE tanımlanmıştır.
XGEVA®, solid tümörlerden kaynaklanan kemik metastazı bulunan hastalarda SRE gelişimi ve çoklu SRE (ilk ve takip edenler) gelişimi riskini azaltmıştır (bkz. Tablo 2).
Tablo 2. Kemik tutulumlu ilerlemiş maligniteleri bulunan hastalarda etkililik sonuçları
| Çalışma 1 meme kanseri | Çalışma 2 diğer solid tümörler** ya da multipl miyelom | Çalışma 3 prostat kanseri | (birleştirilmiş analiz) İleri evre kanser | ||||
| XGEVA | zoledronik asit | XGEVA | zoledronik asit | XGEVA | zoledronik asit | XGEVA | zoledronik asit |
N | 1.026 | 1.020 | 886 | 890 | 950 | 951 | 2.862 | 2.861 |
İlk SRE | ||||||||
Medyan zaman (ay) | NR | 26,4 | 20,6 | 16,3 | 20,7 | 17,1 | 27,6 | 19,4 |
Medyan zamanlar arasındaki fark (ay) | NA | 4,2 | 3,5 | 8,2 | ||||
HR (%95 CI) / RRR (%) | 0,82 (0,71-0,95) / 18 | 0,84 (0,71-0,98) / 16 | 0,82 (0,71-0,95) / 18 | 0,83 (0,76-0,9) / 17 | ||||
Eşit etkililik / Üstünlük p-değerleri | < 0,0001†/ 0,0101†| 0,0007†/ 0,0619†| 0,0002†/ 0,0085†| < 0,0001 / < 0,0001 | ||||
Gönüllülerin oranı (%) | 30,7 | 36,5 | 31,4 | 36,3 | 35,9 | 40,6 | 32,6 | 37,8 |
İlk ve takip eden SRE* | ||||||||
Ortalama sayı/hasta | 0,46 | 0,6 | 0,44 | 0,49 | 0,52 | 0,61 | 0,48 | 0,57 |
Oran (%95 CI) / RRR (%) | 0,77 (0,66-0,89) / 23 | 0,9 (0,77-1,04) / 10 | 0,82 (0,71-0,94) / 18 | 0,82 (0,75-0,89) / 18 | ||||
Üstünlük p-değeri | 0,0012†| 0,1447†| 0,0085†| < 0,0001 | ||||
Yıllık SMR | 0,45 | 0,58 | 0,86 | 1,04 | 0,79 | 0,83 | 0,69 | 0,81 |
İlk SRE ya da HCM | ||||||||
Medyan zaman (ay) | NR | 25,2 | 19 | 14,4 | 20,3 | 17,1 | 26,6 | 19,4 |
HR (%95 CI) / RRR (%) | 0,82 (0,7-0,95) / 18 | 0,83 (0,71-0,97) / 17 | 0,83 (0,72-0,96) / 17 | 0,83 (0,76-0,9) / 17 | ||||
Üstünlük p-değeri | 0,0074 | 0,0215 | 0,0134 | < 0,0001 | ||||
İlk kemik radyasyonu | ||||||||
Medyan zaman (ay) | NR | NR | NR | NR | NR | 28,6 | NR | 33,2 |
HR (%95 CI) / RRR (%) | 0,74 (0,59-0,94) / 26 | 0,78 (0,63-0,97) / 22 | 0,78 (0,66-0,94) / 22 | 0,77 (0,69-0,87) / 23 | ||||
Üstünlük p-değeri | 0,0121 | 0,0256 | 0,0071 | < 0,0001 | ||||
CI = güven aralığı; NR = ulaşılamamıştır; NA = mevcut değil; HCM = malignite hiperkalsemisi; SMR = iskelet morbidite oranı; HR = Risk Oranı; RRR = Rölatif Risk Azalması †1, 2 ve 3 numaralı çalışmalar için ayarlanmış p-değerleri sunulmuştur (ilk SRE ve ilk ve takip eden SRE sonlanım noktaları);
* Zaman içinde görülen tüm iskelet olaylarını açıklar; yalnızca bir önceki olaydan ≥ 21 gün sonra görülen olaylar sayılmıştır.
** NSCLC, renal hücreli kanser, kolorektal kanseri, küçük hücreli akciğer kanseri, mesane kanseri, baş ve boyun kanseri, GI/genitoüriner kanser ve diğerleri dahil, meme ve prostat kanseri hariç.
Şekil 1. Zaman - çalışma sırasındaki ilk SRE için Kaplan-Meier çizimleri
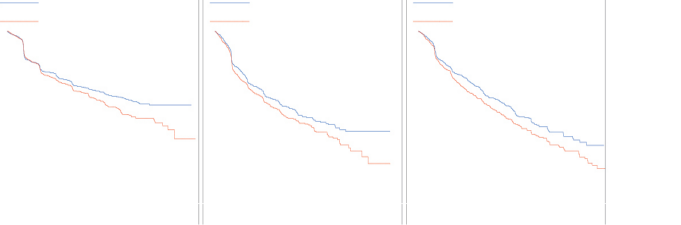
| |||||||||||||||||||
| |||||||||||||||||||
|
|
|
|
|
|
|
|
GRH2312TR v1
Solid tümörlerden metastaz oluşmuş hastalığın ilerlemesi ve genel sağkalım
Hastalığın ilerlemesi, bu üç çalışmada ve üç çalışmanın önceden tanımlanmış birleştirilmiş
analizinde XGEVA® ve zoledronik asit için benzer bulunmuştur.
Kemik tutulumlu ilerlemiş maligniteleri bulunan hastalarda genel sağkalım değerleri, XGEVA® ve zoledronik asit için 1, 2 ve 3 çalışmalar arasında dengelenmiştir: Meme kanseri bulunan hastalar (risk oranı ve %95 CI: 0,95 [0,81-1,11]), prostat kanseri bulunan hastalar (risk oranı ve %95 CI: 1,03 [0,91-1,17]) ve diğer solid tümörleri ya da multipl miyelomu bulunan hastalar (risk oranı ve %95 CI: 0,95 [0,83-1,08]). 2 numaralı çalışma (diğer solid tümörleri ya da multipl miyelomu bulunan hastalar) için yapılan bir post-hoc analizde, kademelendirme için kullanılan 3 tümör tipiyle (küçük hücreli dışı akciğer kanseri, multipl miyelom ve diğer) ilgili genel sağkalım değerleri incelenmiştir. Genel sağkalım, küçük hücreli dışı akciğer kanserinde XGEVA® için daha uzun (risk oranı [%95 CI]: 0,79 [0,65-0,95]; n = 702), multipl miyelomda zoledronik asit için daha uzun (risk oranı [%95 CI]: 2,26 [1,13-4,5]; n = 180) ve diğer tümör türleri için XGEVA® ve zoledronik asit arasında benzerdir (risk oranı [%95 CI]: 1,08 (0,9-1,3); n = 894). Bu çalışmada prognoz faktörleri ve anti-neoplastik tedaviler kontrol edilmemiştir. 1, 2 ve 3 numaralı çalışmalardan alınan önceden tanımlanmış kombine analizde, XGEVA® ve zoledronik asit için genel sağkalım oranları benzer bulunmuştur (risk oranı ve %95 CI 0,99 [0,91-1,07]).
Ağrı üzerindeki etkisi
Ağrıda iyileşmeye kadar geçen süre (BPI-SF en kötü ağrı skorunda başlangıca göre 2 puan ve üstünde azalma), denosumab ve zoledronik asit için her çalışmada ve entegre analizde benzerdir. Birleşik veri setinin post-hoc bir analizinde, ağrının kötüleşmesine kadar geçen medyan süre (> 4 puan en kötü ağrı skoru), başlangıçta hafif ağrısı olan ya da hiç ağrısı olmayan hastalar için XGEVA® için zoledronik aside kısayla daha geçtir (198 güne karşılık 143 gün) (p = 0,0002).
Multipl miyelom hastalarında klinik etkililik
XGEVA®, yeni tanı almış multipl miyelom hastalarında XGEVA® ile zoledronik asidin karşılaştırıldığı uluslararası, randomize (1:1), çift kör, aktif kontrollü çalışma olan çalışma 4'te değerlendirilmiştir.
Bu çalışmada, en az bir kemik lezyonu olan 1.718 multipl miyelom hastası 4 haftada bir (Q4W) subkütan yoldan 120 mg XGEVA® veya 4 haftada bir intravenöz yoldan (IV) 4 mg zoledronik asit (böbrek fonksiyonuna göre ayarlanmış doz) almak üzere randomize edilmiştir. Primer sonuç ölçütü, çalışmadaki ilk iskeletle ilişkili olaya (SRE) kadar geçen süre açısından zoledronik asit ile benzer etkililiğin kanıtlanması olarak belirlenmiştir. Sekonder sonuç ölçütleri arasında ilk SRE'ye kadar geçen süre açısından üstünlük, ilk ve sonraki SRE'ye kadar geçen süre açısından üstünlük ve genel sağkalım yer almıştır. SRE aşağıdakilerden herhangi biri olarak tanımlanmıştır: patolojik kırık (vertebral veya vertebra dışı), kemiğe yönelik radyasyon tedavisi (radyoizotop kullanımı dahil), kemik ameliyatı veya omurilik sıkışması.
Her iki çalışma kolunda, hastaların %54,5'inde otolog PBSC transplantasyonu planlanmış,
%95,8'ine birinci basamak tedavide yeni bir anti-miyelom ajanı uygulanmış/uygulanması planlanmış (yeni tedaviler arasında bortezomib, lenalidomid veya talidomid yer almaktadır) ve %60,7'sinde önceden SRE gelişmiştir. Her iki çalışma kolunda tanı sırasında ISS evre I, evre II ve evre III hasta oranı sırasıyla %32,4, %38,2 ve %29,3 olarak kaydedilmiştir.
Uygulanan medyan doz sayısı XGEVA® için 16, zoledronik asit için 15 olarak bildirilmiştir.
Çalışma 4'ün etkililik bulguları Şekil 2 ve Tablo 3'te sunulmuştur.
Şekil 2. Yeni tanı almış multipl miyelom hastalarında çalışmadaki ilk SRE'ye kadar geçen süre için Kaplan-Meier grafiği

|
|
|
|
|
Tablo 3. Yeni tanı almış multipl miyelom hastalarında zoledronik aside kıyasla XGEVA'ya ilişkin etkililik bulguları
| XGEVA(N = 859) | Zoledronik Asit (N = 859) |
İlk SRE | ||
SRE gelişen hasta sayısı (%) | 376 (43,8) | 383 (44,6) |
SRE'ye kadar geçen medyan zaman (ay) | 22,8 (14,7-NE) | 23,98 (16,56-33,31) |
Risk oranı (%95 CI) | 0,98 (0,85-1,14) | |
| ||
İlk ve sonraki SRE | ||
Ortalama olay/hasta sayısı | 0,66 | 0,66 |
Oran (%95 CI) | 1,01 (0,89-1,15) | |
Yıllık iskeletle ilişkili morbidite oranı | 0,61 | 0,62 |
| ||
İlk SRE veya HCM | ||
Medyan zaman (ay) | 22,14 (14,26-NE) | 21,32 (13,86-29,7) |
Risk oranı (%95 CI) | 0,98 (0,85-1,12) | |
| ||
Kemiğe yönelik ilk radyasyon | ||
Risk oranı (%95 CI) | 0,78 (0,53-1,14) | |
| ||
Genel sağkalım | ||
Risk oranı (%95 CI) | 0,9 (0,7-1,16) | |
NE = tahmin edilemeyen
HCM = malignite hiperkalsemisi
Dev hücreli kemik tümörü bulunan yetişkinlerde ve iskeleti olgunlaşmış adolesanlarda klinik etkililik ve güvenlilik
XGEVA®'nın güvenliliği ve etkililiği, rezeke edilemeyen kemiğin dev hücreli tümörü bulunan veya cerrahi müdahalenin şiddetli morbidite ile ilintili olacağı 554 hastanın kaydedildiği iki adet Faz II açık etiketli, tek kollu çalışmada (çalışma 5 ve 6) incelenmiştir.
Hastalara 8. ve 15. günlerde 120 mg yükleme dozuyla subkütan yoldan 4 haftada bir 120 mg XGEVA® verilmiştir. XGEVA® kullanmayı bırakan hastalar daha sonra minimum 60 ay süren güvenlilik takibi fazına girmiştir. Başlangıçta XGEVA®'ya yanıt veren hastalara güvenlilik takibi sırasında XGEVA® ile yeniden tedaviye izin verilmiştir (örn. hastalık rekürrensi durumunda).
Çalışma 5'e, rezeke edilemez olduğu histolojik olarak doğrulanmış veya nükseden dev hücreli kemik tümörü bulunan 37 yetişkin hasta alınmıştır. Çalışmanın temel sonuç ölçütü, dev hücrelerde başlangıca göre en az %90 eliminasyon (veya dev hücrelerin tümör hücrelerinin
< %5'ini oluşturduğu olgularda dev hücrelerin tam eliminasyonu) ya da histopatolojinin bulunmadığı olgularda radyografik ölçümlere göre hedef lezyonda progresyon olmaması olarak tanımlanan yanıt oranıdır.
Etkililik analizine dahil edilen 35 hastanın %85,7'si (%95 CI: 69,7-95,2), XGEVA®'ya tedavi yanıtı vermiştir. Histoloji değerlendirmesi yapılan 20 hastanın 20'si de (%100) yanıt kriterlerini karşılamıştır. Kalan 15 hastadan 10'unun (%67) radyografik ölçümlerinde hedef lezyonda hiçbir progresyon görülmemiştir.
Çalışma 6'ya dev hücreli kemik tümörü bulunan 535 yetişkin veya iskelet olgunluğuna erişmiş adolesan alınmıştır. Bu hastaların 28'inin 12-17 yaş grubunda olduğu kaydedilmiştir. Hastalar üç kohorttan birine atanmıştır: 1. kohort, cerrahi olarak düzeltilemeyen hastalık bulunan hastaları içermiştir (örn. sakral, spinal veya pulmoner metastazlar dahil çoklu lezyonlar); 2. kohortta cerrahi olarak düzeltilebilecek ancak planlanan ameliyatın şiddetli morbiditeyle ilişki olduğu hastalar yer almıştır (örn. eklem rezeksiyonu, ekstremite ampütasyonu veya hemipelvektomi); 3. kohort, önceden Çalışma 5'te yer alan ve bu çalışmaya geçiş yapan hastaları içermiştir. Primer amaç, dev hücreli kemik tümörü bulunan hastalarda denosumabın güvenlilik profilinin değerlendirilmesidir. Çalışmanın sekonder sonuç ölçütleri arasında 1. kohort için hastalık progresyonuna kadar geçen süre (araştırmacının değerlendirmesine göre) ve 2. kohort için 6. aya kadar ameliyat yapılmayan hastaların oranı yer almıştır.
Nihai analizde, 1. kohortta tedavi alan 260 hastanın 28'inde (%10,8) hastalık progresyonu kaydedilmiştir. İkinci kohortta XGEVA® tedavisi alan değerlendirilebilir 238 hastanın 219'unda (%92 ; %95 CI: %87,8, %95,1) 6. aya kadar ameliyat yapılmadığı görülmüştür. İkinci kohortta yer alan ve başlangıçtaki veya çalışma sırasındaki hedef lezyon yeri akciğer ya da yumuşak doku olmayan 239 hastanın 82'sinde (%34,3) çalışma döneminde ameliyattan kaçınma mümkün olmuştur. İskelet olgunluğuna erişmiş adolesanlardaki genel etkililik bulgularının yetişkinlerde gözlemlenen bulgulara benzer olduğu kaydedilmiştir.
Ağrı üzerindeki etkisi
Kohort 1 ve 2'nin birlikte ele alındığı son analizde, risk altında olan hastaların (yani, başlangıçta en kötü ağrı skoru ≥ 2 olan hastalar) %30,8'inde, tedavinin başlamasından itibaren 1 hafta içinde, en kötü ağrıda klinik açıdan anlamlı bir düşüş (yani, başlangıca göre
≥ 2 puanlık azalma) bildirilmiştir, 5. haftada da ≥ %50'lik bir azalma bildirilmiştir. Bu ağrı iyileşmeleri, sonraki tüm değerlendirmelerde sürmüştür.
Pediyatrik popülasyon
Çalışma 6'da, XGEVA® dev hücreli kemik tümörü bulunan ve en az 1 uzun kemik (örn. humerus epifiz büyüme plağının kapanması) ile vücut ağırlığının 45 kg ve üzerinde olması doğrultusunda tanımlanan iskelet olgunluğuna ulaşmış 28 adolesan (13-17 yaş grubu) hastanın oluşturduğu bir alt kümede değerlendirilmiştir. Cerrahi olarak düzeltilemeyen hastalık bulunan (N=14) bir adolesan hastada, başlangıç tedavisi sırasında hastalık rekürrensi gelişmiştir. Cerrahi olarak düzeltilebilecek hastalık bulunan ancak planlanan ameliyatın şiddetli morbiditeyle ilişkilendirildiği 14 hastanın on üçünün 6. aya kadar ameliyat olmadığı belirlenmiştir.
5.2. Farmakokinetik özellikler
Genel özelliklerEmilim
Subkütan uygulama sonrasında biyoyararlanım %62'dir.
Dağılım
Her 4 haftada bir, birden fazla 120 mg subkütan doz alan kemik metastazı olan kanser hastalarında serum denosumab konsantrasyonunda 2 kata kadar birikim gözlenmiştir.
Biyotransformasyon
Denosumab yalnızca amino asitlerden ve doğal immunoglobulin halinde karbonhidratlardan oluşmuştur ve hepatik metabolik mekanizmalarla elimine edilmesi mümkün değildir. Metabolizması ve eliminasyonunun immunoglobulin klerensi yollarını takip etmesi ve küçük peptitlerle tekil amino asitlere bozunmayla sonuçlanması beklenmektedir.
Eliminasyon
Kanseri ilerlemiş ve her 4 haftada bir, birden fazla 120 mg çoklu dozlama alan gönüllülerde serum denosumab konsantrasyonlarında yaklaşık 2 kata kadar birikim gözlenmiştir ve zamandan bağımsız farmakokinetik ile uyumlu olarak 6 ayda kararlı duruma ulaşılmıştır. Dört haftada bir 120 mg alan multipl miyelom hastalarında medyan dip düzeyler 6. ay ile
12. ay arasında %8'den az değişiklik göstermiştir. Dev hücreli kemik tümörü bulunan, her 4 haftada bir 120 mg olmak üzere 8. ve 15. günlerde bir yükleme dozu alan gönüllülerde, tedavinin ilk ayının içinde kararlı durum düzeylerine ulaşılmıştır. 9. hafta ve 49. hafta arasında, medyan dip düzeyleri %9'dan daha düşük değişkenlik göstermiştir. 4 haftada bir 120 mg dozunu bırakan gönüllülerde, ortalama yarılanma ömrü 28 gündür (14 gün - 55 gün aralığında).
Yapılan bir popülasyon farmakokinetik analizi, durağan haldeki sistemik denosumab maruziyeti için yaş (18 - 87 yaş), ırk/etnik köken (Siyahlar, Hispanikler, Asyalılar ve Beyaz Irk incelenmiştir), cinsiyet ya da solid tümör türleri ya da multipl miyelomlu hastaların bakımından klinik olarak anlamlı bir değişiklik göstermemiştir. Vücut ağırlığı artışı sistemik maruziyetteki azalmayla ilişkilendirilmiştir ve bu durumun tersi de geçerlidir. Kemik döngüsü belirteçlerini baz alan farmakodinamik etkiler geniş bir vücut ağırlığı aralığında tutarlı olduğundan, değişimler klinik açıdan anlamlı bulunmamıştır.
Doğrusallık/doğrusal olmayan durum
Denosumab, geniş bir doz aralığında doza göre doğrusal olmayan bir farmakokinetik sergilemiş, ancak 60 mg (ya da 1 mg/kg) ve üstü dozlar için maruziyette yaklaşık olarak doz orantılı artışlar sergilemiştir. Doğrusal olmayan durum, büyük ihtimalle düşük konsantrasyonlarda önemli olan bir doyurulabilir hedef-aracılı eliminasyon yolundan kaynaklanmaktadır.
Hastalardaki karakteristik özellikler
Böbrek yetmezliği:
İlerlemiş kanseri bulunmayan ancak diyaliz hastaları dahil farklı derecelerde böbrek işlevleri bulunan hastalarda denosumab ile yapılan çalışmalarda (60 mg, n = 55 ve 120 mg, n = 32) böbrek yetmezliğinin denosumabın farmakokinetiğine bir etkisi olmadığı görülmüştür; dolayısıyla böbrek yetmezliği için doz ayarlaması gerekli değildir. XGEVA® tedavisi sırasında böbrek işlevlerinin izlenmesine gerek yoktur.
Karaciğer yetmezliği:
Karaciğer yetmezliği bulunan gönüllülerde özel bir çalışma yapılmamıştır. Genel olarak, monoklonal antikorlar hepatik metabolik mekanizmalarla elimine edilmezler. Denosumabın farmakokinetiğinin karaciğer yetmezliğinden etkilenmesi beklenmemektedir.
Pediyatrik popülasyon:
Dev hücreli kemik tümörü bulunan, 8. ve 15. günlerde yükleme dozuyla birlikte 4 haftada bir 120 mg alan, iskelet olgunluğuna erişmiş adolesanlarda (12-17 yaş grubu) denosumabın farmakokinetik özelliklerinin dev hücreli kemik tümörü bulunan yetişkinlerde görülen farmakokinetik özelliklerle benzer olduğu kaydedilmiştir.
Geriyatrik popülasyon:
Yaşlı hastalarla genç hastalar arasında güvenlilik ya da etkililik bakımından toplamda bir fark gözlemlenmemiştir. Kemik tutulumlu ilerlemiş maligniteleri bulunan 65 yaş üstü hastalarla yapılan kontrollü XGEVA® çalışmalarında, yaşlı ve genç hastalar için benzer etkililik ve güvenlilik bulguları elde edilmiştir. Yaşlı hastalarda doz ayarlaması gerekmemektedir.
5.3. Klinik öncesi güvenlilik verileri
Denosumabın hayvanlardaki biyolojik aktivitesi insan harici primatlara spesifik olduğundan, denosumabın kemirgen modellerindeki farmakodinamik özelliklerini değerlendirebilmek için genetik mühendislik ürünü farelerin (knockout) değerlendirmesine ya da OPG-Fc ve RANK-Fc gibi diğer biyolojik RANK/RANKL yolu inhibitörlerinin kullanımına başvurulmuştur.
OPG-Fc; östrojen reseptörü pozitif ve negatif insan meme kanseri, prostat kanseri ve küçük hücreli dışı akciğer kanseri bulunan fare kemik metastazı modellerinde, osteolitik, osteoblastik ve osteolitik/osteoblastik lezyonları azaltmış, de novo kemik metastazı oluşumunu ertelemiş ve iskelet tümörü büyümesi azaltmıştır. Bu modellerde OPG-Fc hormon tedavisiyle (tamoksifen) ya da kemoterapiyle (dosetaksel) kombine edildiğinde, sırasıyla meme ve prostat ya da akciğer kanserinde iskelet tümör büyümesinde ek inhibisyon görülmüştür. RANK-Fc, bir fare meme tümörü indüksiyon modelinde, meme epitelyumunda hormon kaynaklı proliferasyonu azaltmış ve tümör oluşumunu ertelemiştir.
Denosumabın genotoksisite potansiyelini araştırmak için yapılan standart testler, bu molekül için geçerli olmadıklarından dolayı değerlendirilmemiştir. Bununla birlikte, denosumabın özelliklerinden dolayı bir genotoksisite potansiyeli bulunması çok mümkün değildir.
Denosumabın karsinojenik potansiyeli, uzun vadeli hayvan çalışmalarında değerlendirilmemiştir.
Sinomolgus maymunlarıyla yapılan tek ya da tekrarlı doz toksisitesi çalışmalarında, insanlar için tavsiye edilen dozun 2,7 ila 15 katı sistemik maruziyetle sonuçlanan denosumab dozlarının kardiyovasküler fizyolojiye ve erkek ya da dişi fertilitesine bir etkisi olmamış ve spesifik hedef organ toksisitesi yaratmamıştır.
Gebeliğin ilk trimesterine eşdeğer bir dönem boyunca denosumab dozu verilen sinomolgus maymunlarıyla yapılan bir çalışmada, insanlar için tavsiye edilen dozun 9 katı sistemik maruziyetle sonuçlanan denosumab dozlarının maternal toksisiteye neden olmadığı ve fetal lenf nodları incelenmemiş olmakta birlikte ilk trimestere eşdeğer dönem boyunca fetal zarara neden olmadığı görülmüştür.
Gebelik boyunca insan dozunun 12 katı sistemik maruziyet seviyelerinde denosumab dozu uygulanan sinomolgus maymunlarıyla yapılan bir başka çalışmada, ölü doğum ve postnatal
mortalite oranlarında artış; düşük kemik gücüne neden olan anormal kemik büyümesi, düşük hematopoiez ve diş yer değişimi; periferal lenf nodlarında eksiklik ve neonatal büyümede azalma görülmüştür. Üreme etkileri konusunda ‘hiç advers etki gözlemlenmemiştir' seviyesi elde edilememiştir. Doğumdan sonraki 6 ayın ardından, kemikle ilişkili değişimlerde düzelme görülmüştür ve diş sürmesinde bir etki ortaya çıkmamıştır. Bununla birlikte, lenf nodları ve diş yer değişimi üzerindeki etkiler devam etmiş ve bir hayvanda çeşitli dokularda minimal ila orta dereceli mineralizasyon görülmüştür (tedavi ile ilişkisi belirsiz). Doğum öncesinde maternal zarara ilişkin bir kanıta rastlanmamıştır; doğum sırasında sık olmayan advers maternal etkiler ortaya çıkmıştır. Maternal meme bezi gelişimi normaldir.
Uzun süreli denosumab tedavisi alan maymunlarla yapılan klinik öncesi kemik kalitesi çalışmalarında, kemik döngüsündeki azalmalar kemik gücünde ve normal kemik histolojisindeki iyileşmelerle ilişkilendirilmiştir.
huRANKL'yi eksprese edecek şekilde genetik mühendisliğiyle üretilen ve transkortikal kırığa maruz bırakılan erkek farelerde (knock-in fareler), denosumab kartilajın alınmasını ve kırık kalusunun yeniden şekillenmesini kontrol grubuna kıyasla ertelemiş, ancak biyomekanik güç olumsuz şekilde etkilenmemiştir.
Klinik öncesi çalışmalarda, RANK ve RANKL eksikliği bulunan knockout farelerde, meme bezi matürasyonunun (gebelik sırasında lobulo-alveolar bez gelişimi) inhibisyonu nedeniyle laktasyon görülmemiş ve lenf nodu formasyonunda bozulma görülmüştür. Neonatal RANK/RANKL knockout farelerinde, vücut ağırlığında azalma, kemik büyümesinde azalma, büyüme plaklarında değişim ve diş sürmesinde eksiklik görülmüştür. Düşük kemik büyümesi, büyüme plaklarında değişim ve diş sürmesinde eksiklik, aynı zamanda RANKL inhibitörleri uygulanan neonatal sıçanlarla yapılan çalışmalarda da görülmüştür ve bu değişimler RANKL inhibitörü dozu kesitliğinde kısmen geri döndürülebilir niteliktedir. Klinik maruziyetin 2,7 ila 15 katı (10 ve 50 mg/kg doz) denosumab dozu verilen adolesan primatlarda anormal büyüme plakları görülmüştür. Bu nedenle, denosumab tedavisi büyüme plakları açık olan çocuklarda kemik büyümesine zarar verebilir ve diş çıkarmayı engelleyebilir.
6. FARMASÖTİK ÖZELLİKLER
6.1. Yardımcı maddelerin listesi
Asetik asit (saf)*
Sodyum hidroksit (pH ayarlaması için)*
Sorbitol (E420) Polisorbat 20 Enjeksiyonluk su
* Asetat tamponu, asetik asit ve sodyum hidroksit karıştırılarak hazırlanır
6.2. Geçimsizlikler
Geçimlilik çalışmaları bulunmadığından, bu tıbbi ürün diğer tıbbi ürünlerle karıştırılmamalıdır.
6.3. Raf ömrü
36 ay.
XGEVA®, orijinal kabında saklanmalı ve oda sıcaklığına (25°C) getirildikten sonra 30 gün içinde kullanılmalıdır. XGEVA®, buzdolabından çıkarıldığında 30 gün içinde kullanılmalıdır.
6.4. Saklamaya yönelik özel tedbirler
2°C - 8°C arasında buzdolabında saklayınız. Dondurmayınız.
Işıktan korumak için orijinal ambalajında saklayınız.
6.5. Ambalajın niteliği ve içeriği
Stoper (floropolimer kaplı elastomer) ve yalıtım (alüminyum) ve geçmeli bir kapağa sahip tek kullanımlık flakon içinde (tip I cam) 1,7 ml çözelti.
Bir ve dört flakonluk ambalajlar.
6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemler
XGEVA® çözeltisi, uygulanmadan önce görsel olarak kontrol edilmelidir. Çözelti eser miktarda şeffaf ile beyaz arası renkte proteinli parçacık içerebilir. Çözelti bulanıksa ya da rengi değişmişse enjekte etmeyiniz. Çalkalamayınız. Enjeksiyon bölgesinde rahatsızlık oluşmasını önlemek için, enjekte etmeden önce flakonu oda sıcaklığına (25°C'ye kadar) ısınmaya bırakınız ve yavaş yavaş enjekte ediniz. Flakonun içeriğinin tamamını enjekte ediniz. Denosumabın uygulanması için 27 numara bir iğne tavsiye edilir. Flakona birden fazla kez iğne sokmayınız.
Kullanılmamış olan ürünler ya da atık materyaller ‘Tıbbi Atıkların Kontrolü Yönetmeliği' ve
‘Ambalaj ve Ambalaj Atıklarının Kontrolü Yönetmelikleri'ne uygun olarak imha edilmelidir.
 Grip, Soğuk Algınlığı ve Öksürük
Grip ve soğuk algınlığı (nezle) semptomları arasındaki farkı bilmek önemlidir. Soğuk algınlığı gripten daha hafif belirtiler gösteren bir solunum yolu hastalığıdır.
Grip, Soğuk Algınlığı ve Öksürük
Grip ve soğuk algınlığı (nezle) semptomları arasındaki farkı bilmek önemlidir. Soğuk algınlığı gripten daha hafif belirtiler gösteren bir solunum yolu hastalığıdır. |
 Sırt Ağrısı
Sırt ağrısı birden bire ortaya
çıkıp şiddetli (akut) olabilir veya zamanla gelişip daha uzun
süreli sorunlara (kronik) neden olabilir.
Sırt Ağrısı
Sırt ağrısı birden bire ortaya
çıkıp şiddetli (akut) olabilir veya zamanla gelişip daha uzun
süreli sorunlara (kronik) neden olabilir. |
İLAÇ EŞDEĞERLERİ
| Eşdeğer İlaç Adı | Barkodu | İlaç Fiyatı |
|---|---|---|
| XGEVA | 8699862770019 | 5,397.18TL |
| Diğer Eşdeğer İlaçlar |
 |
Kalp Krizi Kalbe giden kan akışı durduğunda kalp krizi meydana gelir. |
 |
Ruh ve Akıl Sağlığımızı Geliştirmek İyi akıl ve ruh sağlığı sahip olmaktan ziyade, yaptığınız şeylerdir. Akıl ve ruhsal olarak sağlıklı olmak için kendinize değer vermeli ve kendinizi kabul etmelisiniz. |
 |
Mide Kanseri Mide kanseri genellikle mideyi tümüyle kaplayan ve mukus üretmekle görevli hücrelerde başlar. Bu kanser tipine adenokarsinom denir. |
İLAÇ GENEL BİLGİLERİ
Amgen İlaç Tic. Ltd. Şti
| Geri Ödeme Kodu | A16763 |
| Satış Fiyatı | 5397.18 TL [ 26 Apr 2024 ] |
| Önceki Satış Fiyatı | 5397.18 TL [ 22 Apr 2024 ] |
| Original / Jenerik | Original İlaç |
| Reçete Durumu | Normal Reçeteli bir ilaçdır. |
| Barkodu | 8699862770019 |
| Etkin Madde | Donesumab |
| ATC Kodu | M05BX04 |
| Birim Miktar | 120 |
| Birim Cinsi | MG |
| Ambalaj Miktarı | 1 |
| Kas İskelet Sistemi > Kemik İlaçları > Denosumab |
| İthal ( ref. ülke : Fransa ) ve Beşeri bir ilaçdır. |