XELJANZ XR 11 mg uzat�lm�� sal�ml� film kapl� tablet(28 tablet) K�sa �r�n Bilgisi
{ Tofacitinib }
1. BE�ER� TIBB� �R�N�N ADI
XELJANZXR 11 mg uzat�lm�� sal�ml� film kapl� tablet
2. KAL�TAT�F VE KANT�TAT�F B�LE��M
Etkin madde
Her uzat�lm�� sal�ml� film kapl� tablet, 11 mg tofasitinibe e�de�er 17,771 mg tofasitinib sitrat i�erir.
Yard�mc� maddeler
Her uzat�lm�� sal�ml� film kapl� tablet 152,229 mg sorbitol (E420) i�ermektedir. Yard�mc� maddeler i�in b�l�m 6.1'e bak�n�z.
3. FARMAS�T�K FORMU
Uzat�lm�� sal�ml� film kapl� tablet.
Oval, pembe, bir ucu delikli, uzat�lm�� sal�ml� film kapl� bir tablettir.
4. KL�N�K �ZELL�KLER
4.1. Terap�tik endikasyonlar
Romatoid artrit
XELJANZ XR, eri�kinlerde orta veya �iddetli aktif romatoid artritte (RA) bir veya daha fazla TNF blokeri kullan�m�na yetersiz cevap olmas� veya intolerans olmas� durumunda endikedir.
XELJANZ XR di�er JAK inhibit�rleri, biyolojik DMARD'lar, azatioprin ve siklosporin gibi potent imm�ns�presiflerle birlikte kullan�lmas� uygun de�ildir.
Ps�riatik artrit
XELJANZ XR eri�kinlerde aktif ps�riatik artrit (PsA) tedavisinde bir veya daha fazla TNF blokeri kullan�m�na yetersiz cevap olmas� veya intolerans olmas� durumunda endikedir.
XELJANZ XR, di�er JAK inhibit�rleri, biyolojik DMARD'lar, azatioprin ve siklosporin gibi potent imm�ns�presiflerle birlikte kullan�lmas� uygun de�ildir.
Ankilozan spondilit
XELJANZ XR, eri�kinlerde ankilozan spondilit (AS) tedavisinde bir veya daha fazla TNF blokeri kullan�m�na yetersiz cevap olmas� veya intolerans olmas� durumunda endikedir.
XELJANZ XR biyolojik anti-romatizmal ila�larla (DMARD) veya azatiyopirin ve siklosporin gibi potent imm�ns�presiflerle birlikte kullan�lmas� uygun de�ildir.
4.2. Pozoloji ve uygulama �ekli
Kullan�m ile ilgili �nemli talimatlar
11 mg uzat�lm�� sal�ml� film kapl� tablet ile 5 mg film kapl� tablet aras�ndaki ge�i� hekim taraf�ndan yap�lmal�d�r.
Mutlak lenfosit say�s� <500 h�cre/ mm'�n alt�nda, mutlak n�trofil say�s� (ANC)
<1000 h�cre/ mm alt�nda ve hemoglobin seviyeleri 9 g/dL'nin alt�nda olan
hastalarda tofasitinib ile tedaviye ba�lanmamal�d�r.
Lenfopeni, n�tropeni ve aneminin kontrol alt�na al�nmas�nda doz kesilmesi �nerilir (bkz. b�l�m 4.4 ve 4.8).
Ciddi enfeksiyon geli�en hastalarda enfeksiyon kontrol alt�na al�nana kadar
tofasitinib kullan�m� kesilmelidir (bkz. b�l�m 4.4).
XELJANZ XR, yemeklerle birlikte veya tek ba��na al�nmal�d�r (bkz. b�l�m 5.1).
XELJANZ XR b�t�n olarak yutulmal�d�r. Ezilmemeli, par�alanmamal� ya da
�i�nenmemelidir.
XELJANZ XR tedavisi, XELJANZ XR'�n endike oldu�u durumlar�n te�his ve tedavisinde deneyimli uzman hekimler taraf�ndan ba�lat�lmal� ve izlenmelidir.
Romatoid artrit, ps�riatik artrit ve ankilozan spondilit
A�a��daki tablo 1 XELJANZ XR'in RA, PsA ve AS endikasyonlar�nda yeti�kin hastalardaki g�nl�k doz �nerisini ve CYP2C19 ve/veya CYP3A4 inhibit�r� alan hastalarda, orta veya �iddetli b�brek yetmezli�i olan hastalarda (hemodiyalize giren �iddetli yetmezlik hastalar�n� da i�erir ama bunlarla s�n�rl� de�ildir) veya orta �iddette karaci�er yetmezli�i olan hastalarda ve lenfopeni, n�tropeni ve anemi durumlar�ndaki doz �nerilerini g�stermektedir.
Yeti�kin hastalarda tavsiye edilen doz, g�nde bir kez uygulanan 11 mg uzat�lm�� sal�ml� film kapl� tablettir. Bu doz a��lmamal�d�r.
Tablo 1: RA, PsAve AS hastalar�nda XELJANZ XR i�in doz �nerileri
| XELJANZ XR uzat�lm�� sal�ml� film kapl� tablet |
Yeti�kin hastalar | G�nde bir kez 11 mg |
inhibit�r�(leri) ile beraber g��l� CYP2C19 inhibit�r� (�rn flukonazol) kullanan hastalar (bkz ila� etkile�imleri) | RA ve PsA endikasyonu i�in doz g�nde bir kez 5 mg film kapl� tablete d���r�lmelidir AS endikasyonu i�in doz her iki g�nde bir 11 mg uzat�lm�� sal�ml� tablet olarak devam edilmelidir. |
(bkz. �zel pop�lasyonlar ile ilgili ek bilgiler) olan hastalar (bkz. �zel pop�lasyonlar ile ilgili ek bilgiler) | RA ve PsA endikasyonu i�in doz g�nde bir kez 5 mg film kapl� tablete d���r�lmelidir. AS endikasyonu i�in doz her iki g�nde bir 11 mg uzat�lm�� sal�ml� tablet olarak devam edilmelidir. |
| Hemodiyalize giren hastalarda doz diyaliz uygulamas�ndan sonra ayn� g�n verilmelidir. E�er diyaliz prosed�r�nden �nce doz al�nd� ise diyaliz sonras� ek doz �nerilmez. |
Tekrarlanan testlerle lenfosit say�s�n�n <500 h�cre/ mm'�n alt�nda oldu�u do�rulanan hastalar | Tedavi kesilir. |
ANC de�eri 500-1000 h�cre/mm olan hastalar | Tedavi kesilir. ANC>1,000 h�cre/mm oldu�unda g�nde tek doz 11 mg ile devam edilir. |
ANC<500 h�cre/mm olan hastalar | Tedavi kesilir. |
Hemoglobin <8,0 g/dL veya 2 g/dL'den fazla d���� | Hemoglobin de�erleri normalle�ene kadar uygulamaya ara verilir. |
G��l� CYP3A4 inhibit�r� (�rn. ketokonazol) veya
Orta derecede CYP3A4
Orta veya �iddetli b�brek yetmezli�i
Orta derecede karaci�er yetmezli�i
XELJANZ XR'�n �iddetli karaci�er yetmezli�inde kullan�m� �nerilmez.
11 mg uzat�lm�� sal�ml� film kapl� tablet ile 5 mg film kapl� tablet aras�ndaki ge�i�
G�nde 2 kez 5 mg tablet kullanan hastalar 5 mg dozlar�n� ald�klar� g�n� takip eden g�nde, g�nde 1 kez XELJANZ XR 11 mg uzat�lm�� sal�ml� tablet kullanmaya ba�layabilirler.
Uygulama �ekli:
XELJANZ XR, oral yoldan yemeklerle birlikte veya tek ba��na al�nabilir.
XELJANZ XR 11 mg uzat�lm�� sal�ml� film kapl� tablet, dozun do�ru miktarda al�nd���ndan emin olunabilmesi i�in b�t�n olarak yutulmal�d�r. Ezilmemeli, par�alanmamal� ya da �i�nenmemelidir.
�zel pop�lasyonlara ili�kin ek bilgiler:
B�brek yetmezli�i:
Orta ve �iddetli yetmezlik
XELJANZ ile tedavi g�ren orta veya �iddetli b�brek yetmezli�i hastalar� normal b�brek fonksiyonu (bkz. b�l�m 5.1) olan XELJANZ hastalar� ile kar��la�t�r�ld���nda daha y�ksek tofasitinib kan konsantrasyonuna sahiptir. Bu nedenle orta veya �iddetli b�brek yetmezli�i olan hastalarda (hemodiyalize giren �iddetli yetmezlik hastalar�n� da i�erir ama bunlarla s�n�rl� de�ildir) XELJANZ XR'�n dozunun ayarlanmas� �nerilmektedir (bkz. pozoloji/uygulama s�kl��� ve s�resi-tablo 1).
Hafif yetmezlik
Hafif b�brek yetmezli�i olan hastalarda doz ayarlmas� gerekli de�ildir.
Karaci�er yetmezli�i:
�iddetli yetmezlik
XELJANZ XR �iddetli karaci�er yetmezli�i hastalar�nda �al���lmam��t�r. Bu nedenle XELJANZ XR'�n �iddetli karaci�er yetmezli�i hastalar�nda kullan�m� �nerilmez.
Orta �iddette yetmezlik
XELJANZ ile tedavi g�ren orta karaci�er yetmezli�i hastalar� normal karaci�er fonksiyonu (bkz. b�l�m 5.1) olan XELJANZ hastalar� ile kar��la�t�r�ld���nda daha y�ksek tofasitinib kan konsantrasyonuna sahiptir. Y�ksek kan konsantasyonu baz� yan etkilerin g�r�lme riskini artt�rabilir. Bu nedenle orta �iddette karaci�er yetmezli�i olan hastalarda XELJANZ XR'�n dozunun ayarlanmas� �nerilmektedir (bkz. Pozoloji/uygulama s�kl��� ve s�resi-tablo 1).
Hafif yetmezlik
Hafif karaci�er yetmezli�i olan hastalarda doz ayarlmas� gerekli de�ildir.
Hepatit B veya C serolojisi
Hepatit B vir�s veya hepatit C vir�s serolojik tan�s� pozitif olan hastalarda XELJANZ XR'�n g�venlili�i veya etkilili�i �al���lmam��t�r.
Pediyatrik pop�lasyon:
Tofasitinibin pediyatrik hastalarda g�venlili�i ve etkilili�i belirlenmemi�tir.
Geriyatrik pop�lasyon:
RA �al��malar�na kat�lan 3315 hastadan 505 RA hastas� 65 ya� ve �zeri olup bunun 71 tanesi 75 ya� ve �zeridir. Tofasitinib ile tedavi edilen hastalarda ciddi enfeksiyonlar�n g�r�lme s�kl��� 65 ya� ve �zeri hastalarda 65 ya� ve alt�ndakilere k�yasla daha y�ksektir.
Genel olarak ya�l� hastalarda enfeksiyon insidans�ndaki y�kseklik nedeniyle bu ya�
grubunu tedavi ederken dikkatli olunmal�d�r (bkz. b�l�m 4.4).
Diyabetik hastalar:
Genel olarak ya�l� ve diyabeti olan pop�lasyonda enfeksiyon s�kl��� daha y�ksek oldu�undan, bu gruptaki hastalar�n tedavisi s�ras�nda dikkatli olunmal�d�r.
4.3. Kontrendikasyonlar
Etkin madde
Aktif t�berk�loz (TB), sepsis veya f�rsat�� enfeksiyonlar gibi ciddi enfeksiyonlarda (bkz. b�l�m 4.4).
Ciddi karaci�er yetmezli�inde (bkz. b�l�m 4.2).
Hamilelik ve emzirme d�neminde (bkz. b�l�m 4.6).
4.4. �zel kullan�m uyar�lar� ve �nlemleri
65 ya� ve �zeri hastalarda kullan�m
65 ya� ve �zeri hastalardaki artm�� ciddi enfeksiyon, miyokardiyal enfarkt�s, malignite ve t�m nedenlere ba�l� mortalite riski nedeniyle bu grup hastada tofasitinib ba�ka uygun tedavi alternatifi yoksa kullan�lmal�d�r (daha fazla detay i�in b�l�m 4.4 ve 5.1 ba�l�klar�na bak�n�z).
Mortalite
En az bir ek kardiyovask�ler (KV) risk fakt�r� g�r�len, 50 ya��nda veya daha b�y�k romatoid artrit (RA) hastalar�yla yap�lan geni�, randomize, pazarlama sonras� g�venlilik �al��mas�nda, t�m�r nekroz fakt�r� (TNF) inhibit�rlerine k�yasla g�nde 2 kez 5 mg tofasitinib veya g�nde 2 kez 10 mg tofasitinib ile tedavi edilen hastalarda ani kardiyovask�ler �l�mler dahil olmak �zere t�m nedenlere ba�l� �l�m oran� daha y�ksektir. T�m nedenlere ba�l� �l�mlerin insidans oran� 100 hasta y�l� ba��na g�nde 2 kez 5 mg tofasitinib kullan�m�nda 0,88; g�nde 2 kez 10 mg tofasitinib kullan�m�nda 1,23 ve TNF blokerlerinin kullan�m�nda ise 0,69'dur. Tofasitinib kullan�m�na ba�lamadan veya tedaviye devam etmeden �nce hastalar risk yarar a��s�ndan bireysel olarak de�erlendirilmelidir.
RA, PsA veya AS tedavisinde g�nde 2 kez 10 mg tofasitinib kullan�m� �nerilmez (bkz. b�l�m 4.2).
Di�er ila�larla kombine kullan�m
Tofasitinibin TNF antagonistleri, interl�kin (IL)-1R antagonistleri, IL-6R antagonistleri, anti-CD20 monoklonal antikorlar�, IL-17 antagonistleri, IL-12/IL-23 antagonistleri, anti- integrinler, selektif kostimulan mod�lat�rler ve azatiopirin, 6-merkaptop�rin, siklosporin ve takrolimus gibi g��l� immuns�presan tedavilerle beraber kullan�lmas� denenmemi�tir ve y�ksek immuns�presyon ve artm�� enfeksiyon riski nedeniyle �nerilmemektedir.
Romatoid artrit klinik �al��malar�nda, tofasitinibin MTX kombinasyonu ile g�r�len advers olaylar�n insidans� tofasitinib monoterapisine k�yasla daha y�ksektir.
Tofasitinibin fosfodiesteraz 4 inhibit�rleri ile kombinasyon halinde kullan�m� klinik olarak �al���lmam��t�r.
Tromboz
�nflamatuvar ko�ullar� tedavi etmek i�in tofasitinib ve di�er Janus kinaz (JAK) inhibit�rleri ile tedavi edilen hastalarda baz�lar�n�n �l�mle sonu�land��� pulmoner emboli (PE), derin damar trombozu (DVT) ve arteriyel trombozun da dahil oldu�u ciddi tromboz olaylar� g�r�lm��t�r. Bu olaylar�n bir�o�u ciddi olup baz�lar� �l�mle sonu�lanm��t�r.
50 ya� ve �st� ve en az bir ek kardiyovask�ler risk fakt�r�ne sahip RA'l� hastalarda yap�lan ruhsatland�rma sonras� g�venlilik �al��mas� 1'de, TNF inhibit�r� alan hastalara k�yasla g�nde 2 kez 5 mg ve g�nde 2 kez 10 mg tofasitinib kullananlarda tromboz olaylar�n�n insidans�nda art�� g�zlenmi�tir (b�l�m 4.8 ve 5.1'e bak�n�z). DVT insidans oran� 100 hasta y�l� ba��na g�nde 2 kez 5 mg tofasitinib kullan�m�nda 0,22; g�nde 2 kez 10 mg tofasitinib kullan�m�nda 0,28 ve TNF blokerlerinin kullan�m�nda ise 0,16'd�r. PE insidans oran� 100 hasta y�l� ba��na g�nde 2 kez 5 mg tofasitinib kullan�m�nda 0,18; g�nde 2 kez 10 mg tofasitinib kullan�m�nda 0,49 ve TNF blokerlerinin kullan�m�nda ise 0,05'dir.
RA, PsA ve AS tedavisinde g�nde 2 kez 10 mg tofasitinib kullan�m� �nerilmez (bkz. b�l�m 4.2).
�al��ma sonras� g�zlemsel analizde bilinen tromboz risk fakt�rleri olan hastalarda tekrar trombozun ortaya ��kmas� tofasitinib alan hastalarda 12. ay D-dimer seviyesi ≥2× ULN olanlarda D-dimer seviyesi <2×ULN olanlara g�re daha y�ksek ��km��t�r, bu risk TNF inhibit�rleri ile tedavi edilen hastalarda belirgin de�ildir. Az say�daki tromboz olaylar� ve s�n�rl� D-Dimer testi yap�lm�� olmas� (yaln�zca ba�lang��ta, 12. ayda ve �al��ma bitiminde de�erlendirilmi�tir) nedeniyle s�n�rl� yorum yap�labilmektedir. �al��ma boyunca tromboz geli�meyen hastalarda, 12. ayda t�m tedavi kollar�nda ortalama D-dimer seviyesi ba�lang�� seviyesine g�re anlaml� �l��de azalm��t�r. Ancak hastalar�n yakla��k %30'unda 12. ayda D-dimer seviyesi ≥2× ULN olmas�na kar��n tromboz olaylar� g�zlenmemi�tir. Bu durum bu �al��madaki D-dimer testinin duyarl�l���n�n s�n�rl� oldu�unu g�stermektedir.
Tromboz i�in bilinen risk fakt�rleri olan RA'l� hastalar i�in yakla��k 12 ayl�k tedaviden sonra D-dimer seviyesinin test edilmesi d���n�lmelidir.
D-dimer test seviyesi ≥2× ULN ise tofasitinib ile tedaviye devam etme karar�ndan �nce klinik yararlar�n risklerden fazla oldu�u do�rulanmal�d�r.
MACE veya malignite risk fakt�rleri olan hastalarda (bkz. b�l�m 4.4 “Major advers kardiyovask�ler olaylar” ve “Malignite ve lenfoproliferatif hastal�k”) tofasitinib yaln�zca uygun tedavi alternatifleri yok ise kullan�lmal�d�r.
MACE veya malignite risk fakt�rleri d���ndaki tromboz risk fakt�rlerine sahip hastalarda tofasitinib dikkatli kullan�lmal�d�r. MACE veya malignite risk fakt�rleri d���ndaki tromboz risk fakt�rleri; daha �nceden ge�irilmi� tromboz, major bir ameliyat ge�irecek olmas�, immobilizasyon, kombine hormonal kontraseptifler veya hormon replasman tedavisi kullan�m�, kal�t�msal koag�lasyon bozuklu�u i�erir Hastalar�n tofasitinib tedavisi boyunca tromboz risk fakt�rlerinde de�i�im i�in periyodik olarak tekrar de�erlendirilmesi �nerilir.
Doza veya endikasyona bak�lmaks�z�n tromboz belirti ve bulgular� olan hastalar hemen de�erlendirilmeli ve tromboz belirtileri olan hastalarda tofasitinibe devam edilmemelidir.
Tofasitinib tromboz riski artm�� olan hastalarda kullan�lmamal�d�r. Retinal ven�z tromboz
Retinal ven�z tromboz (RVT) tofasitinib ile tedavi edilen hastalarda raporlanm��t�r (bkz. b�l�m 4.8). RVT'yi i�aret eden herhangi bir durumu tecr�be eden hastalara derhal medikal yard�m almalar� tavsiye edilmelidir.
Ciddi enfeksiyonlar
Tofasitinib tedavisi alan hastalarda bakteriyel, mikobakteriyel, invazif fungal, viral ya da di�er f�rsat�� patojenlere ba�l� ciddi ve bazen fatal enfeksiyonlar bildirilmi�tir (bkz. b�l�m 4.8). Tofasitinib ile bildirilen en yayg�n ciddi enfeksiyonlar aras�nda, pn�moni, sel�lit, herpes zoster, idrar yolu enfeksiyonu, divertik�lit ve apandisittir. F�rsat�� enfeksiyonlar aras�ndan t�berk�loz ve di�er mikobakteriyel enfeksiyonlar, kriptokok, histoplasmoz, �zofageal kandidiyaz, pn�mosistoz, multidermatomal herpes zoster, sitomegalovir�s, BK vir�s� enfeksiyonlar� ve listeriyoz tofasitinib kullan�m� ile bildirilmi�tir. Baz� hastalar, lokalize yerine yay�lm�� enfeksiyon g�stermi�tir ve genellikle metotreksat veya kortikosteroid gibi imm�nomod�lat�r ajanlar ile birlikte kullanm��lard�r.
F�rsat�� enfeksiyon riski Asya co�rafi b�lgelerinde daha y�ksektir. Kortikosteroid alan romatoid artrit hastalar� enfeksiyona yatk�n olabilir.
Tofasitinib, lokal enfeksiyonlar da dahil olmak �zere aktif enfeksiyonu olan hastalarda kullan�m�ndan ka��n�lmal�d�r.
A�a��daki durumlarda tofasitinibe ba�lamadan �nce tedavinin hastalar a��s�ndan faydalar� ve riskleri g�z �n�nde bulundurulmal�d�r.
Kronik veya tekrarlay�c� enfeksiyonlar� olan hastalar,
T�berk�loza maruz kalm�� hastalar,
Ciddi ya da f�rsat�� enfeksiyon ge�mi�i olan hastalar,
Endemik t�berk�loz veya endemik mikoz bulunan yerlerde ya�am�� ya da buralara seyahat etmi� olan hastalar,
Enfeksiyona yatk�nl��a neden olabilecek altta yatan ko�ullar� bulunan hastalar
Tofasitinib ile tedavi s�ras�nda ya da tedaviden sonra hastalar enfeksiyon belirtileri ve semptomlar� a��s�ndan yak�ndan izlenmelidir. Hastada ciddi bir enfeksiyon, f�rsat�� enfeksiyon ya da sepsis ortaya ��kmas� halinde ilaca ara verilmelidir. Tofasitinib tedavisi s�ras�nda yeni bir enfeksiyon g�r�len bir hastaya, imm�nitesi zay�flam�� bir hasta i�in gerekli, tan�sal testler derhal ve eksiksiz olarak yap�lmal�, uygun antimikrobiyal tedavi ba�lamal� ve hasta yak�ndan izlenmelidir.
Genel olarak ya�l� ve diyabeti olan pop�lasyonda enfeksiyon s�kl��� daha y�ksek oldu�undan, bu gruptaki hastalar�n tedavisi s�ras�nda dikkatli olunmal�d�r (bkz. b�l�m 4.8). 65 ya� ve �zerindeki hastalarda tofasitinib sadece ba�ka uygun tedavi alternatifleri yok ise kullan�lmal�d�r (bkz. b�l�m 5.1).
Lenfopeni derecesindeki art�� ile enfeksiyon riski daha y�ksek olabilir ve hastalardaki enfeksiyon riskini de�erlendirirken lenfosit say�s�na dikkat edilmelidir. Tedavinin durdurulmas� ve lenfopeni kriterlerinin g�zlenmesi b�l�m 4.2'de tart���lm��t�r.
T�berk�loz
A�a��daki durumlarda tofasitinibe ba�lamadan �nce tedavinin hastalar a��s�ndan faydalar� ve riskleri g�z �n�nde bulundurulmal�d�r.
T�berk�loza maruz kalm�� hastalar,
Endemik t�berk�loz bulunan yerlerde ya�am�� ya da buralara seyahat etmi� olan
hastalar.
Tofasitinib tedavisinden �nce ve tedavi s�resince periyodik olarak hastalar latent ya da aktif enfeksiyon a��s�ndan uygun k�lavuzlara g�re de�erlendirilmeli ve tetkik edilmelidir.
Yeterli tedavi alm�� oldu�u do�rulanamayan, TB testi negatif latent ya da aktif TB ge�mi�i olan hastalarda veya testi negatif oldu�u halde TB enfeksiyonu a��s�ndan risk fakt�rleri bulunan hastalar i�in tofasitinib tedavisinden �nce anti-t�berk�loz tedavi gereklili�i de�erlendirilmelidir. TB tedavisinde deneyimli bir uzman taraf�ndan yap�lan kons�ltasyon ile hastalar�n anti-t�berk�loz tedavisi i�in uygunluk karar�n�n verilmesi �nerilmektedir.
Tedaviye ba�lamadan �nce latent TB enfeksiyonu testi negatif ��kan hastalar da dahil olmak �zere hastalar TB belirtileri ve semptomlar�n�n geli�imi a��s�ndan yak�ndan izlenmelidir.
Latent TB enfeksiyonu olan hastalar tofasitinib uygulamas� �ncesi standart bir antimikobakteriyel ajan ile tedavi edilmelidir.
Viral reaktivasyon
Tofasitinib ile yap�lan klinik �al��malarda herpes vir�s reaktivasyonu (�rne�in herpes zoster) dahil olmak �zere viral reaktivasyon g�zlenmi�tir (bkz.b�l�m 4.8).
Pazarlama sonras� ara�t�rmalarda tofasitinib ile tedavi g�ren hastalarda hepatit B reaktivasyonu bildirilmi�tir. Tofasitinibin kronik viral hepatit reaktivasyonu �zerindeki etkisi bilinmemektedir. Tarama s�ras�nda Hepatit B ya da C i�in pozitif sonu� veren hastalar, klinik �al��malardan ��kar�lm��t�r. Tofasitinib ile tedaviye ba�lanmadan �nce klinik k�lavuzlar do�rultusunda viral hepatit i�in tarama yap�lmal�d�r.
Tofasitinib ile tedavi edilen a�a��daki hasta gruplar�nda herpes zoster insidans�nda art�� g�r�lm��t�r.
Japon ve Koreli hastalar
ALC 1,000 h�cre/mm'den az olan hastalarda (bkz. b�l�m 4.2),
Daha �nceden 2 veya daha fazla biyolojik DMARD alan uzun s�reli RA hastalar�nda,
G�nde 2 kere 10 mg kullanan hastalarda
Major advers kardiyovask�ler olaylar
50 ya� ve �st� ve en az bir ek kardiyovask�ler risk fakt�r�ne sahip RA'l� hastalarda yap�lan g�venlilik �al��mas� 1'de, TNF inhibit�r� alan hastalara k�yasla g�nde 2 kez 5 mg ve g�nde
2 kez 10 mg tofasitinib kullananlarda kardiyovask�ler �l�m, �l�mc�l olmayan miyokardiyal enfarkt�s (MI) ve �l�mc�l olmayan inme olarak tan�mlanan maj�r advers kardiyovask�ler olaylar�n (MACE) oran�nda art�� olmu�tur. MACE insidans oran� 100 hasta y�l� ba��na g�nde 2 kez 5 mg tofasitinib kullan�m�nda 0,91; g�nde 2 kez 10 mg tofasitinib kullan�m�nda 1,11 ve TNF blokerlerinin kullan�m�nda ise 0,79'dur. �l�mc�l veya �l�mc�l olmayan miyokardiyal enfarkt�s insidans oran� 100 hasta y�l� ba��na g�nde 2 kez 5 mg tofasitinib kullan�m�nda 0,36; g�nde 2 kez 10 mg tofasitinib kullan�m�nda 0,39 ve TNF blokerlerinin kullan�m�nda ise 0,20'dir. Ge�mi�te ya da halen sigara i�en hastalar ek risk alt�ndad�r.
65 ya� ve �zerinde halen veya ge�mi�te uzun s�redir sigara i�en hastalarda ve aterosklerotik kardiyovask�ler hastal�k �yk�s� veya di�er kardiyovask�ler risk fakt�rleri olan hastalarda tofasitinib yaln�zca uygun tedavi alternatifi yok ise kullan�lmal�d�r (bkz b�l�m 5.1).
RA veya PsA tedavisinde g�nde 2 kez 10 mg tofasitinib kullan�m� �nerilmez (bkz. b�l�m 4.2).
Maligniteler ve lenfoproliferatif hastal�k
Tofasitinib ki�inin malignitelere kar�� savunma mekanizmas�n� etkileyebilir.
Tofasitinib ile yap�lan klinik �al��malarda lenfomalar ve kat� t�m�rler dahil olmak �zere maligniteler g�zlenmi�tir (bkz. b�l�m 4.8).
RA g�venlilik �al��mas� 1'de TNF inhibit�r� alan hastalara k�yasla g�nde 2 kez 5 mg veya g�nde 2 kez 10 mg tofasitinib ile tedavi edilen hastalarda daha y�ksek oranda malignite (�zellikle NMSC) oran� g�zlenmi�tir. Malignitelerin insidans oran� 100 hasta y�l� ba��na g�nde 2 kez 5 mg tofasitinib kullan�m�nda 1,13; g�nde 2 kez 10 mg tofasitinib kullan�m�nda 1,13 ve TNF blokerlerinin kullan�m�nda ise 0,77'dir. Ge�mi�te ya da halen sigara i�en hastalar ek risk alt�ndad�r.
RA g�venlilik �al��mas� 1'de t�m malignitelerin bir alt k�mesi olan lenfomalar ve akci�er kanserleri TNF inhibit�r� alan hastalara k�yasla g�nde 2 kez 5 mg veya g�nde 2 kez 10 mg tofasitinib ile tedavi edilen hastalarda daha y�ksek oranda g�r�lm��t�r. Lenfomalar�n insidans oran� 100 hasta y�l� ba��na g�nde 2 kez 5 mg tofasitinib kullan�m�nda 0,07; g�nde 2 kez 10 mg tofasitinib kullan�m�nda 0,11 ve TNF blokerlerinin kullan�m�nda ise 0,02'dir. Ge�mi�te ya da halen sigara i�en hastalar aras�nda akci�er kanserlerinin insidans oran� 100 hasta y�l� ba��na g�nde 2 kez 5 mg tofasitinib kullan�m�nda 0,48; g�nde 2 kez 10 mg tofasitinib kullan�m�nda 0,59 ve TNF blokerlerinin kullan�m�nda ise 0,27'dir.
Tofasitinib kullan�m�na ba�lamadan veya tedaviye devam etmeden �nce �zellikle bilinen malignitesi olan (ba�ar�l� bir �ekilde tedavi edilen NMSC d���nda), tedavi s�ras�nda malignite geli�tiren ve ge�mi�te uzun s�re i�mi� ya da halen sigara i�en hastalar risk yarar a��s�ndan bireysel olarak de�erlendirilmelidir. RA veya PsA tedavisinde g�nde 2 kez 10 mg tofasitinib kullan�m� �nerilmez (bkz. b�l�m 4.2).
De novo b�brek nakli hastalar�nda yap�lan kontroll� doz-aral�kland�rma �al��malar�nda (Faz 2B), t�m� basiliksimab, y�ksek doz kortikosteroidler ve mikofenolik asit �r�nleri ile ind�ksiyon tedavisi alan 218 tofasitinib hastas�n�n (%2,3) 5 tanesinde Epstein Barr Vir�s� ile ili�kili nakil sonras� lenfoproliferatif bozukluk g�zlemlenirken 111 siklosporin hastas�n�n hi�birinde bu bozukluk g�zlenmemi�tir.
Tofasitinib ile tedavi edilen hastalarda akci�er kanseri, meme kanseri, melanom, prostat kanseri ve pankreas kanseri dahil fakat bunlarla s�n�rl� olmamak �zere klinik �al��malarda ve pazarlama sonras� ara�t�rmalarda di�er maligniteler g�zlenmi�tir.
Melanom d��� cilt kanseri
T�m hastalar i�in ama �zellikle cilt kanseri i�in artm�� risk ta��yan hastalar i�in periyodik cilt muayenesi �nerilmektedir (bkz. b�l�m 4.8).
�nterstisyel akci�er hastal���
Kronik akci�er hastal��� hikayesi olan hastalarda veya interstisyel akci�er hastal��� geli�tirmi� hastalarda enfeksiyona daha yatk�n olmalar� nedeniyle dikkatli olunmas� �nerilir. RA klinik �al��malar�nda ve pazarlama sonras�nda tofasitinib kullanan hastalarda interstisyel akci�er hastal��� (baz�lar� �l�m ile sonu�lanm��t�r) raporlanm��t�r. Janus kinaz (JAK) inhibisyonunun bu olaylardaki rol� bilinmemektedir. Asyal� RA hastalar�n�n interstisyel akci�er hastal��� a��s�ndan daha y�ksek risk alt�nda oldu�u bilindi�inden bu hastalar�n tedavisinde dikkatli olunmal�d�r.
Gastrointestinal perforasyon
Tofasitinib ile y�r�t�len klinik �al��malarda gastrointestinal perforasyon olgular� rapor edilmi�tir ancak bu olaylarda JAK inhibisyonunun rol� bilinmemektedir.
Gastrointestinal perforasyon riski artm�� olan (�rne�in divertik�lit ge�mi�i, e� zamanl� kortikosteroid ve/veya non steroidal antiinflamatuvar ila� kullan�m�) hastalarda tofasitinib dikkatli bir �ekilde kullan�lmal�d�r. Yeni ba�layan abdominal belirti ve bulgular ile hastaneye ba�vuran hastalar, gastrointestinal perforasyonunun erken tan�s� i�in derhal de�erlendirilmelidir (bkz. b�l�m 4.8).
K�r�klar
Tofasitinib alan hastalarda k�r�k geli�imi g�zlenmi�tir.
Tofasitinib ileri ya�ta hastalar, kad�n hastalar ve kortikosteroid kullanan hastalar gibi bilinen k�r�k risk fakt�rleri olan hastalarda doz ve endikasyondan ba��ms�z dikkatli kullan�lmal�d�r.
Karaci�er enzimlerinde art��
Tofasitinib ile tedavi plasebo ile kar��la�t�r�ld���nda karaci�er enzimlerinde y�kselme insidans�nda art�� ile ili�kilendirilmi�tir. Bu anormalliklerin �o�u �ncesinde DMARD (�ncelikle metotreksat) tedavisi olan �al��malarda meydana gelmi�tir.
Y�ksek alanin aminotransferaz (ALT) veya aspartat aminotransferaz (AST) seviyeleri olan hastalarda tofasitinib tedavisi �zellikle MTX gibi potansiyel hepatotoksik ila�larla birlikte ba�lat�lmas� d���n�l�yorsa dikkatli olunmal�d�r.
Potansiyel ilaca ba�l� karaci�er hasar� olgular�n� belirleyebilmek i�in karaci�er testleri rutin olarak takip edilmeli ve karaci�er enzimlerindeki y�kselmenin sebepleri h�zl�ca ara�t�r�lmal�d�r. E�er ilaca ba�l� karaci�er hasar�ndan ��phelenilirse, bu te�his ortadan kalkana kadar tofasitinib tedavisine ara verilmelidir.
Hipersensitivite
Tofasitinib kullanan hastalarda ilaca a��r� duyarl�l���n i�areti olabilen anjiyo�dem ve �rtiker gibi reaksiyonlar g�zlenmi�tir. Baz� olaylar ciddidir. Ciddi hipersensitivite reaksiyonunun g�r�lmesi durumunda potansiyel neden veya reaksiyona neyin sebep oldu�u de�erlendirilirken tofasitinib tedavisi derhal kesilmelidir (bkz. b�l�m 4.8).
Laboratuvar anomalileri Lenfosit anomalileri
Tofasitinib tedavisi plasebo ile kar��la�t�r�ld���nda lenfopeni insidans�nda art�� ile ili�kilendirilmi�tir. 750 h�cre/mm'�n alt�ndaki lenfosit say�lar� ciddi enfeksiyon insidans�nda art�� ile ili�kilendirilmi�tir. Lenfosit sat�s� 750 h�cre/mm'�n alt�nda olan hastalarda tofasitinib tedavisine ba�lanmas� veya devam edilmesi �nerilmez.
Lenfosit seviyesi ba�lang��ta ve ard�ndan her 3 ayda bir izlenmelidir. Lensofit say�s� baz al�narak �nerilen d�zenlemeler i�in b�l�m 4.2'ye bak�n�z.
N�tropeni
Tofasitinib tedavisi, plasebo ile kar��la�t�r�ld���nda artm�� n�tropeni (<2,000 h�cre/mm) insidans� ile ili�kilendirilmi�tir. ANC'si 1.000 h�cre/mm'�n alt�nda olan hastalarda tofasitinib tedavisine ba�lanmas� �nerilmez.
Tedavi ba�lang�c�nda, 4-8 hafta sonra ve ard�ndan her 3 ayda bir ANC izlenmelidir. ANC baz al�narak �nerilen d�zenlemeler i�in b�l�m 4.2'ye bak�n�z.
Anemi
Tofasitinib tedavisi, hemoglobin seviyelerinde d����ler ile ili�kilendirilmi�tir. Hemoglobin de�eri d���k olan hastalarda (<9 g/dL) hastalarda tofasitinib tedavisinden ka��n�lmal�d�r.
Hemoglobin seviyesi; ba�lang��ta, tedavi ba�lang�c�ndan 4-8 hafta sonra ve ard�ndan her 3 ayda bir izlenmelidir. Hemoglobin seviyesi baz al�narak �nerilen d�zenlemeler i�in b�l�m
4.2'ye bak�n�z.
Lipid de�erlerinde art��
Tofasitinib tedavisi; total kolesterol, d���k yo�unluklu lipoprotein (LDL) kolesterol ve y�ksek yo�unluklu lipoprotein (HDL) kolesterol gibi lipid parametrelerindeki art��lar ile ili�kilendirilmi�tir. Maksimum etkiler genellikle 6 hafta i�inde g�zlenmi�tir. Tofaistinib tedavisinin ba�lamas�ndan 8 hafta sonra lipid parametrelerinin de�erlendirilmesi yap�lmal�d�r.
Hastalar, hiperlipidemi tedavisine y�nelik klinik k�lavuzlara (uluslararas� kolesterol e�itim program� (NCEP)) g�re tedavi edilmelidir.
Tofasitinib tedavisi ile ili�kili olan toplam kolesterol ve LDL kolesterol seviyelerindeki art�� statin tedavisi ile tofasitinib �ncesi seviyelere d���r�lebilir.
Diyabet tedavisi g�ren hastalarda hipoglisemi
Diyabet tedavisi g�ren hastalarda tofasitinib ba�lanmas�n� takiben hipoglisemi raporlanm��t�r. Hipogliseminin g�r�lmesi durumunda anti-diyabetik ila�larda doz ayarlamas� gerekebilir.
A��lamalar
Tofasitinib tedavisine ba�lamadan �nce mevcut ba����klama k�lavuzlar�na uygun olarak ba����klamalar� g�ncelleyiniz.
Canl� a��lar�n, tofasitinib ile birlikte uygulanmas�ndan ka��n�n�z. Tofasitinib tedavisinden �nce canl� a�� uygulama karar� verilirken, hastada var olan imm�nos�presyon dikkate al�nmal�d�r.
Profilaktik zoster a��s� a��lama k�lavuzlar�na g�re de�erlendirilmelidir. Daha �nce iki veya daha fazla biyolojik DMARD alm��, uzun s�reli RA'l� hastalara �zellikle dikkat edilmelidir.
Canl� zoster a��s� yap�l�yorsa; sadece su �i�e�i �yk�s� olan veya varicella zoster vir�s� (VZV) i�in seropozitif olan hastalara uygulanmal�d�r. Su �i�e�i ge�mi�i ��pheli veya g�venilir de�ilse, VZV'ye kar�� antikorlar�n test edilmesi �nerilir. Canl� a��larla a��lama, tofasitinib ba�lang�c�ndan en az 2 hafta, tercihen 4 hafta �nce veya imm�nomod�lat�r t�bbi �r�nlerle ilgili g�ncel a��lama k�lavuzlar�na uygun olarak yap�lmal�d�r. Tofasitinib alan hastalara canl� a��lar yoluyla enfeksiyonun sekonder bula�mas�na ili�kin herhangi bir veri mevcut de�ildir.
Deforme olmayan uzat�lm�� sal�m form�lasyonu ile gastrointestinal obstr�ksiyon riski Hali haz�rda ciddi gastrointestinal daralma (patalojik yada iatrojenik) problemi olan hastalarda tofasitinib uzat�lm�� sal�ml� film kapl� tablet uygularken dikkatli olunmal�d�r. Bozulmayan bir uzat�lm�� sal�m form�lasyonu kullanan di�er ila�lar�n yutulmas� ile ba�lant�l� olarak bilinen darl�klara sahip hastalarda obstr�ktif semptomlar nadir olarak bildirilmi�tir.
Yard�mc� maddeler
Bu t�bbi �r�n, her tablette sorbitol (E420) i�erir. Nadir kal�t�msal fruktoz intolerans problemi olan hastalar�n bu ilac� kullanmamalar� gerekir. Sorbitol (veya fruktoz) i�eren �r�nler ile diyetle sorbitol (veya fruktoz) al�m�n�n birlikte uygulanmas�n�n ilave etkisi dikkate al�nmal�d�r.
Oral kullan�lan �r�nlerdeki sorbitol miktar� beraber kullan�ld��� di�er oral ila�lar�n biyoyararlan�m�n� etkileyebilir.
4.5. Di�er t�bbi �r�nler ile etkile�imler ve di�er etkile�im �ekilleri
Di�er ila�lar�n tofasitinibin farmakokineti�ini (PK) etkileme potansiyelleri
Tofasitinib, CYP3A4 ile metabolize edildi�inden, CYP3A4'� inhibe eden ya da ind�kleyen ila�lar ile etkile�imi olabilir. Tofasitinib maruziyeti, potent CYP3A4 inhibit�rleri (ketokonazol) ya da orta derecede CYP3A4 ve potent CYP2C19 (�rn. flukonazol) inhibisyonuna sebep olan bir veya birka� ilac�n beraber kullan�lmas� ile artar (bkz. b�l�m 4.2).
Tofasitinib maruziyeti potent CYP ind�kleyicileri (�rn. rifampin) ile birlikte uyguland���nda azalmaktad�r. Tek ba��na CYP2C19 ya da P-glikoprotein inhibit�rlerinin tofasitinib farmakokineti�ini belirgin bir �ekilde de�i�tirmesi beklenmez.
Ketokonazol (g��l� bir CYP3A4 inhibit�r�), flukonazol (orta derecede bir CYP3A4 ve potent CYP2C19 inhibit�r�), takrolimus (hafif CYP3A4 inhibit�r�) ve siklosporin (orta derecede bir CYP3A4 inhibit�r�) ile birlikte kullan�m� tofasitinib EAA's�n� artt�r�rken
rifampisin (potent CYP ind�kleyicisi) tofasitinib EAA's�n� d���rmektedir. Tofasitinibin potent CYP ind�kleyicileri (�rn. rifampisin) ile birlikte kullan�lmas� klinik cevap al�namamas�na veya klinik cevab�n azalmas�na sebep olabilir (bkz. �ekil 1). Tofasitinibin potent CYP3A4 ind�kleyicileri ile birlikte kullan�lmas� �nerilmemektedir. Ketokonazol ve flukonazol ile birlikte kullan�lmas� tofasitinibin Cde�erini artt�r�rken takrolimus, siklosporin ve rifampisin ile birlikte kullan�lmas� tofasitinibin Cde�erini azaltmaktad�r.
RA hastalar�nda MTX ile e�zamanl� olarak uygulaman�n (haftada bir kez 15-25 mg MTX), tofasitinib farmakokineti�i �zerinde bir etkisi olmam��t�r (bkz. �ekil 1).
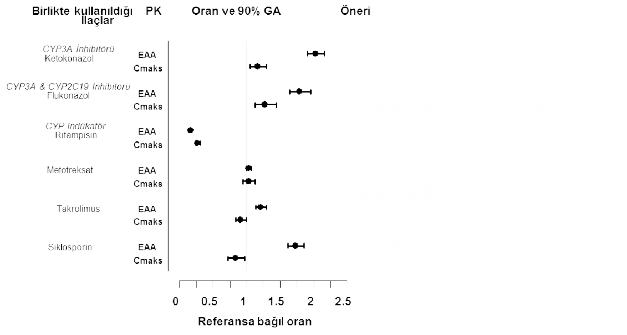
Tofasitinib dozu d���r�lmelidir Tofasitinib dozu d���r�lmelidir
Etkilili�i d���rebilir.
Doz ayarlamas�na gerek yoktur.
Tofasitinibin takrolimus ile birlikte kullan�m�ndan ka��n�lmal�d�r.
Tofasitinibin siklosporin ile birlikte kullan�m�ndan ka��n�lmal�d�r.
�ekil 1. Di�er ila�lar�n tofasitinib farmakokineti�i (PK) �zerine etkisi
Not: Referans gruba tofasitinib tek ba��na uygulanm��t�r.
Tofasitinibin di�er ila�lar�n PK'lar�n� etkileme potansiyeli
Sa�l�kl� kad�n g�n�ll�lerde tofasitinibin birlikte uyguland��� oral kontraseptiflerin
levonorgestrel ve etinil estradiol farmakokineti�i �zerinde bir etkisi olmam��t�r.
RA hastalar�nda tofasitinibin haftada bir kez 15-25 mg MTX ile birlikte uygulanmas�, MTX EAA ve Cde�erlerini s�ras�yla %10 ve %13 oran�nda d���rm��t�r. MTX maruziyetindeki d���� miktar�, bireysel MTX dozunda ayarlama yap�lmas�n� gerektirmemektedir.
�zel pop�lasyonlara ili�kin ek bilgiler Pediyatrik pop�lasyon:
Etkile�im �al��malar� sadece yeti�kinlerde ger�ekle�tirilmi�tir.
4.6. Gebelik ve laktasyon
Gebelik kategorisi : C
�ocuk do�urma potansiyeli bulunan kad�nlar/Do�um kontrol� (Kontrasepsiyon) �ocuk do�urma potansiyeli bulunan kad�nlara tofasitinib tedavisi s�ras�nda ve son dozdan 4 hafta sonras�na kadar etkili do�um kontrol y�ntemleri kullanmalar� �nerilmelidir.
Gebelik d�nemi
Tofasitinibin gebe kad�nlarda kullan�m�na ili�kin yeterli veri mevcut de�ildir. Tofasitinibin s��anlarda ve tav�anlarda teratojenik oldu�u ve do�umu ve peri/postnatal geli�imi etkiledi�i g�sterilmi�tir (bkz. b�l�m 5.3). �nsanlara y�nelik potansiyel risk bilinmemektedir.
�nlem olarak tofasitinib gebelik d�neminde kontrendikedir (bkz. b�l�m 4.3). Risk �zeti
Hamile kad�nlarda XELJANZ XR kullan�m�na dair mevcut veriler, ila� ile ili�kili maj�r do�um defektleri, d���k veya advers maternal ya da fetal sonu� riskinin belirlenmesi i�in yeterli de�ildir. Hamilelik s�ras�nda romatoid artrit ve �lseratif kolit ile ili�kili anne ve fet�s� etkileyen riskler s�z konusudur (bkz. klinik endi�eler). Hayvan �reme �al��malar�nda tofasitinib, maksimum �nerilen doz olan g�nde 2 kez 10 mg dozunun s�ras�yla 73 ve 6,3 kat� maruziyet d�zeyi durumunda, organogenez d�nemindeki hamile s��anlar ve tav�anlarda, fetosidal ve teratojenik etkilere sebep olmu�tur. Ayr�ca s��anlarda ger�ekle�tirilen peri- ve post-natal bir �al��mada tofasitinib, �nerilen g�nl�k 2 kez 5 mg dozun yakla��k 73 kat ve maksimum �nerilen doz olan g�nde 2 kez 10 mg dozun yakla��k 36 kat �zerindeki maruziyet d�zeylerinde, canl� yavru boyutunda, postnatal sa�kal�mda ve yavru v�cut a��rl�klar�nda d����e sebep olmu�tur (bkz. veriler).
�lac�n endike oldu�u pop�lasyonlardaki tahmini maj�r do�um defekti ya da d���k riski bilinmemektedir. T�m gebelikler altta yatan bir do�um kusuru ya da kayb� riski veya di�er advers sonu�lara ait riskler ta��maktayd�r. Genel ABD pop�lasyonunda klinik olarak tan�nm�� olan gebeliklerde maj�r do�um defektleri ve d���kler i�in risk d�zeyi s�ras�yla
%2-4 ve %15-20 d�zeyindedir.
Klinik endi�eler
Hastal�k ile ili�kili maternal ve/veya embriyo/fetal risk
Yay�mlanm�� olan veriler, romatoid artrit ya da �lseratif kolit hastas� kad�nlarda hastal�k aktivitesindeki art���n advers gebelik sonu�lar�n�n geli�imi ile ili�kili oldu�una i�aret etmektedir. Advers gebelik sonu�lar� aras�nda preterm do�um (gestasyonun 37. haftas�ndan
�nce), d���k do�um a��rl�kl� (< 2500 g) ve do�umdaki gestasyonel ya�a k�yasla k���k infantlar yer almaktad�r.
Veriler
Hayvan verileri
Hamile s��anlara organogenez s�ras�nda tofasitinib uygulanan s��an embriyofetal geli�im �al��mas�nda tofasitinibin, g�nl�k �nerilen doz olan g�nde 2 kez 5 mg'�n yakla��k 146 kat �zerindeki ve maksimum �nerilen doz olan g�nde 2 kez 10 mg'�n (s��anlarda 100 mg/kg/g�n oral dozlardaki EAA baz�nda) yakla��k 73 kat �zerindeki maruziyet d�zeylerinde teratojenik oldu�u g�sterilmi�tir.
Teratojenik etkiler aras�nda, s�ras�yla anasarka ve membran�z ventrik�ler septal defektler olmak �zere eksternal ve yumu�ak doku malformasyonlar�; ve iskelet malformasyonlar� veya varyasyonlar� yer almaktad�r (servikal ark eksikli�i; b�k�lm�� femur, fibula, humerus, radius, skapula, tibia ve ulna; sternokizi; kaburga eksikli�i; deforme femur; dallanm�� kaburga, kayna�m�� kaburga; kayna�m�� sternebra; ve hemisentrik torasik merkez). Buna ek olarak, canl� fet�s say�s�nda azalma ile sonu�lanan, erken ve ge� rezorpsiyonlar ile karakterize implantasyon sonras� kay�plarda bir art�� g�zlenmi�tir. Ortalama fetal v�cut a��rl��� d��m��t�r. S��anlarda tofasitinibin, g�nl�k �nerilen doz olan g�nde 2 kez 5 mg'�n yakla��k 58 kat �zerindeki ve maksimum �nerilen doz olan g�nde 2 kez 10 mg'�n (hamile s��anlarda 30 mg/kg/g�n oral dozlardaki EAA baz�nda) yakla��k 29 kat �zerindeki maruziyet d�zeylerinde herhangi bir geli�imsel toksisite g�zlemlenmemi�tir.
Hamile tav�anlara organogenez s�ras�nda tofasitinib uygulanan tav�an embriyofetal geli�im �al��mas�nda tofasitinibin, g�nl�k �nerilen doz olan g�nde 2 kez 5 mg'�n yakla��k 13 kat �zerindeki ve maksimum �nerilen doz olan g�nde 2 kez 10 mg'�n (tav�anlarda 30 mg/kg/g�n oral dozlardaki EAA baz�nda) yakla��k 6,3 kat �zerindeki maruziyet d�zeylerinde maternal toksisiste belirtisi olmaks�z�n teratojenik oldu�u g�sterilmi�tir. Teratojenik etkiler aras�nda; torakogastro�izis, omfalosel, membran�z ventrik�ler septal defektler ve kranial / iskelet malformasyonlar� (mikrostomi, mikroftalmi), orta hat ve kuyruk defektleri yer almaktad�r. Buna ek olarak, ge� rezorpsiyonlar ile ili�kili olarak implantasyon sonras� kay�plarda bir art�� g�zlenmi�tir. Tav�anlarda tofasitinibin, g�nl�k �nerilen doz olan g�nde 2 kez 5 mg'�n yakla��k 3 kat �zerindeki ve maksimum �nerilen doz olan g�nde 2 kez 10 mg'�n (hamile tav�anlarda 10 mg/kg/g�n oral dozlardaki EAA baz�nda) yakla��k 1,5 kat �zerindeki maruziyet d�zeylerinde herhangi bir geli�imsel toksisite g�zlemlenmemi�tir.
Hamile s��anlarda ger�ekle�tirilen peri- ve post-natal bir geli�im �al��mas�nda 6. gestasyon g�n�nden laktasyonun 20. g�n�ne kadar tofasitinib, �nerilen g�nde 2 kez 5 mg dozun yakla��k 73 kat ve maksimum �nerilen doz olan g�nde 2 kez 10 mg dozun (s��anlarda 50 mg/kg/g�n oral dozlarda EAA baz�nda) yakla��k 36 kat �zerindeki maruziyet d�zeylerinde, canl� yavru boyutunda, postnatal sa�kal�mda ve yavru v�cut a��rl�klar�nda d����e sebep olmu�tur. S��anlarda �nerilen g�nde 2 kez 5 mg dozun yakla��k 17 kat� ve maksimum �nerilen doz olan g�nde 2 kez 10 mg dozun (s��anlarda 10 mg/kg/g�n oral dozlardaki EAA baz�nda). yakla��k 8,3 kat� maruziyet d�zeylerinde canl� F2 jenerasyon fet�slerin �retilmesinde ve F1 jenerasyon s��anlar�n davran��sal ve ��renme
de�erlendirmelerinde, seks�el olgunla�mas�nda ya da �iftle�me yetenekleri �zerinde herhangi bir etki saptanmam��t�r.
Laktasyon d�nemi
Risk �zeti
Tofasitinibin insan s�t�ne ge�ip ge�medi�i, emzirilen infant �zerindeki etkileri ya da s�t �retimi �zerindeki etkilerine dair herhangi bir veri bulunmamaktad�r. Tofasitinib emzirmekte olan s��anlar�n s�t�ne ge�mi�tir (bkz. veriler). Emzirme durumunda �ocuk i�in risk d��lanamaz. �nlem olarak tofasitinib emzirme d�neminde kontrendikedir (bkz. b�l�m 4.3).
�reme yetene�i/Fertilite
�nsan do�urganl��� �zerindeki potansiyel etkiye ili�kin resmi �al��malar yap�lmam��t�r. Tofasitinib, s��anlarda di�i fertilitesini bozmu� ancak erkek fertilitesini etkilememi�tir (bkz. b�l�m 5.3).
4.7. Ara� ve makine kullan�m� �zerindeki etkiler
Tofasitinibin ara� ve makine kullanma yetene�i �zerine etkisi yoktur veya ihmal edilebilir d�zeydedir.
4.8. �stenmeyen etkiler
G�venlilik profili �zeti Romatoid artritEn yayg�n g�r�len ciddi advers reaksiyonlar, ciddi enfeksiyonlar olmu�tur (bkz. b�l�m 4.4). T�m maruziyet pop�lasyonunda uzun d�nem g�venlilik verilerine g�re tofasitinib kullan�m� ile en yayg�n bildirilen ciddi enfeksiyonlar pn�moni (%1,7), herpes zoster (%0,6), idrar yolu enfeksiyonlar� (%0,4), sel�lit (%0,4), divertik�lit (%0,3) ve apandisittir (%0,2). F�rsat�� enfeksiyonlar aras�ndan TB ve di�er mikobakteriyel enfeksiyonlar, kriptokok, histoplasmoz, �zofageal kandidiyaz, multidermatomal herpes zoster, sitomegalovir�s, BK vir�s� enfeksiyonlar� ve listeriyoz tofasitinib kullan�m� ile bildirilmi�tir. Baz� hastalar, lokalize yerine yay�lm�� enfeksiyon bulgular� ile ba�vurmu�tur. Klinik �al��malarda bildirilmeyen di�er ciddi enfeksiyonlar da ortaya ��kabilir (�rn. koksidiyoidomikoz).
�ift k�r, plasebo veya MTX kontroll� klinik �al��malar�n ilk 3 ay�nda en yayg�n rapor edilen advers reaksiyonlar; ba� a�r�s� (%3,9), �st solunum yolu enfeksiyonlar� (%3,8), viral �st solunum yolu enfeksiyonlar� (%3,3), diyare (%2,9), mide bulant�s� (%2,7) ve hipertansiyon (%2,2) olmu�tur.
�ift-k�r, plasebo veya MTX kontroll� �al��malar�n ilk 3 ay�nda herhangi bir advers reaksiyona ba�l� olarak tedaviyi b�rakma oran� tofasitinib alan hastalar i�in %3,8'dir. Kontroll� klinik �al��malar�n ilk 3 ay�nda tedavinin b�rak�lmas� ile sonu�lanan en yayg�n enfeksiyonlar; herpes zoster (%0,19) ve pn�monidir (%0,15).
Ps�riatik artrit
Genel olarak, tofasitinib ile tedavi edilen aktif PsA hastalar�nda g�zlenen g�venlilik profili
tofasitinib ile tedavi edilen RA hastalar�nda g�zlenen g�venlilik profili ile uyumludur.
Ankilozan spondilit
Genel olarak, XELJANZ XR ile tedavi edilen AS hastalar�nda g�zlenen g�venlilik profili XELJANZ ile tedavi edilen RA ve PsA hastalar�nda g�zlenen g�venlilik profili ile uyumludur.
RA, PsA ve AS hastalar�nda yap�lan klinik �al��malara g�re advers ila� reaksiyonlar�, sistem/organ s�n�f� ve a�a��da belirtilen sisteme g�re belirlenmi� s�kl�k kategorisine g�re s�n�fland�r�lm��t�r; �ok yayg�n (≥1/10); yayg�n (≥1/100 ila <1/10); yayg�n olmayan (≥1/1.000 ila <1/100); seyrek (≥1/10.000 ila <1/1.000); �ok seyrek (< 1/10.000); bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor). Her s�kl�k grubunda, istenmeyen etkiler azalan ciddiyet s�ras�na g�re yer almaktad�r.
Enfeksiyonlar ve enfestasyonlar
Yayg�n : | Pn�moni, influenza, herpes zoster, idrar yolu enfeksiyonu, sin�zit, bron�it, nazofarenjit, farenjit |
Yayg�n olmayan : | T�berk�loz, divertik�lit, piyelonefrit, sel�lit, herpes simpleks, viral gastroenterit, viral enfeksiyon |
Seyrek : | Sepsis, �rosepsis, dissemine t�berk�loz, bakteriyemi, Pn�mosistis jiroveci pn�monisi, pn�mokokal pn�moni, bakteriyel pn�moni, sitomegalovir�s enfeksiyonu, bakteriyel artrit |
�ok seyrek | Merkezi sinir sistemi t�berk�lozu, kriptokokal menenjit, nekrotizan fasit, ensefalit, stafilokokal bakteremi Mikobakteriyum avium kompleks enfeksiyonu, atipik mikobakteriyel enfeksiyon |

Yayg�n olmayan : Akci�er kanseri, melanom d��� cilt kanseri Seyrek : Lenfoma
(Kist ve polipler de dahil olmak �zere) iyi huylu ve k�t� huylu neoplazmalar
Kan ve lenf sistemi hastal�klar�
Yayg�n : | Lenfopeni, anemi |
Yayg�n olmayan : | L�kopeni, n�tropeni |
Ba����kl�k sistemi hastal�klar�
Bilinmiyor : | �laca a��r� duyarl�l�k*, anjiyo�dem*, �rtiker* |
Metabolizma ve beslenme hastal�klar�
Yayg�n olmayan : Dislipidemi, hiperlipidemi, dehidratasyon
Psikiyatrik hastal�klar
Yayg�n olmayan : �nsomnia
Sinir sistemi hastal�klar� Yayg�n : Ba� a�r�s� Yayg�n olmayan : Parestezi
Kardiyak hastal�klar
Yayg�n olmayan : Miyokardiyal enfarkt�s
Vask�ler hastal�klar
Yayg�n : Hipertansiyon
Yayg�n olmayan : Ven�z tromboemboli**
Solunum, g���s bozukluklar� ve mediastinal hastal�klar
Yayg�n : �ks�r�k
Yayg�n olmayan : Dispne, sin�s konjesyonu
Gastrointestinal hastal�klar
Yayg�n : Abdominal a�r�, kusma, diyare, bulant�, gastrit, dispepsi
Hepato-biliyer hastal�klar
Yayg�n olmayan : Hepatik steatoz, hepatik enzim art���, transaminaz art���, gama
-glutamiltransferaz art���,
Seyrek : Anormal karaci�er test sonu�lar�
Deri ve deri alt� doku hastal�klar� Yayg�n : D�k�nt� Yayg�n olmayan : Eritem, pruritus
Kas-iskelet bozukluklar, ba� doku ve kemik hastal�klar�
Yayg�n : Artralji
Yayg�n olmayan : Eklem �i�li�i, tendinit
Seyrek : Muskuloskeletal a�r�
Genel bozukluklar ve uygulama b�lgesine ili�kin hastal�klar
Yayg�n : Periferik �dem Yayg�n olmayan : Pireksi, yorgunluk
Ara�t�rmalar
Yayg�n : Kan kreatin fosfokinaz d�zeyi art���
Yayg�n olmayan : Kan kreatinin art���, kan kolesterol d�zeyi art���, d���k yo�unluklu
lipoprotein art���, kilo art���
Yaralanma ve zehirlenme
Yayg�n olmayan : Ligamentte burkulma, kas gerilmesi
* Spontan raporlanan veri
** Ven�z tromboemboli PE, DVT ve Retinal Ven�z Tromboz'u i�erir.
Se�ilmi� yan etkilerin a��klanmas� Ven�z tromboemboli
Romatoid artrit
En az bir ek kardiyovask�ler (KV) risk fakt�r� g�r�len, 50 ya��nda veya daha b�y�k romatoid artrit hastalar�yla yap�lan geni� (N=4.362), randomize, onay sonras� g�venlilik �al��mas�nda, TNF inhibit�rlerine k�yasla tofasitinib ile tedavi edilen hastalarda doza ba��ml� olarak artan insidansta VTE g�zlenmi�tir (bkz. b�l�m 5.3). Bu olaylar�n �o�u ciddidir ve baz�lar� �l�mle sonu�lanm��t�r. G�nde iki kez 5 mg tofasitinib, g�nde iki kez 10 mg tofasitinib ve TNF inhibit�rleri i�in PE insidans oranlar� (%95 GA) her 100 hasta- y�l i�in s�ras�yla 0,17 (0,08-0,33), 0,50 (0,32-0,74) ve 0,06 (0,01-0,17)'dur. TNF
inhibit�rleri ile kar��la�t�r�ld���nda PE risk oran� (HR), g�nde iki kez 5 mg tofatinib ve g�nde iki kez 10 mg tofasitinib i�in s�ras�yla 2,93 (0,79-10,83) ve 8,26 (2,49-27,43)'dur (bkz. b�l�m 5.1). Tofasitinib ile tedavi edilen ve PE g�r�len hastalar�n �o�unlu�unda (%97) VTE risk fakt�r� bulunmaktad�r.
Ankilozan spondilit
Birle�tirilmi� faz 2 ve faz 3 randomize kontroll� klinik �al��malarda 48 hafta kadar
tofasitinib alan 420 hastada (233 hasta-y�l g�zlem) VTE g�r�lmemi�tir.
Genel enfeksiyonlar
Romatoid artrit
0-3 ay boyunca, kontroll� Faz 3 klinik �al��malar�nda, g�nde iki kez 5 mg (toplam 616 hasta) ve g�nde iki kez 10 mg (toplam 642 hasta) tofasitinib monoterapi gruplar�ndaki enfeksiyon oranlar� s�ras�yla %16,2 (100 hasta) ve %17,9 (115 hasta) iken, plasebo grubunda (toplam 122 hasta) bu oran %18,9 (23 hasta) olmu�tur. DMARD kombinasyon tedavisi i�eren kontroll� Faz 3 klinik �al��malar�nda, g�nde iki kez 5 mg (toplam 973 hasta) ve g�nde iki kez 10 mg (toplam 969 hasta) tofasitinib art� DMARD gruplar�ndaki ilk 3 aydaki enfeksiyon oranlar�, s�ras�yla %21,3 (207 hasta) ve %21,8 (211 hasta) iken plasebo art� DMARD grubunda (toplam 559 hasta) bu oran %18,4 (103 hasta) olmu�tur.
En yayg�n rapor edilen enfeksiyonlar, �st solunum yolu enfeksiyonu ve nazofarenjit olmu�tur (s�ras�yla %3,7 ve %3,2).
Uzun s�reli t�m g�venlilik maruziyet pop�lasyonunda (toplam 4,867 hasta) tofasitinib ile
genel enfeksiyon oran�, 100 hasta y�l� ba��na 46,1 olay olmu�tur (g�nde iki kez 5 mg ve 10
mg i�in s�ras�yla 43,8 ve 47,2 hasta). Monoterapi alan hastalar i�in (toplam 1,750) oran 100 hasta y�l� ba��na g�nde iki kez 5 mg ve 10 mg i�in s�ras�yla 48,9 ve 41,9 olayd�r. DMARD'lar ile kombinasyon tedavisi alan hastalar i�in (toplam 3,117), enfeksiyon oranlar� 100 hasta y�l� ba��na g�nde iki kez 5 mg ve 10 mg i�in s�ras�yla 41,0 ve 50,3 olay olarak ger�ekle�mi�tir.
Ankilozan spondilit
Birle�tirilmi� faz 2 ve faz 3 randomize kontroll� klinik �al��malarda, 16 haftaya kadar ki placebo kontroll� �al��ma periyodunda g�nde 2 kere 5 mg tofasitinib grubunda (185 hasta) enfeksiyonlar�n g�r�me s�kl��� % 27,6 iken placebo grubunda (187 hasta) bu oran %23'd�r. Birle�tirilmi� faz 2 ve faz 3 randomize kontroll� klinik �al��malarda 48 haftaya kadar g�nde iki kez tofasitinib 5 mg alan 316 hastada enfeksiyon g�r�lme oran� % 35,1'dir.
Ciddi enfeksiyonlar
Romatoid artrit
6 ay ve 24 ay s�reli 2 kontroll� klinik �al��mada, g�nde iki kez 5 mg tofasitinib monoterapisi grubunda g�zlenen ciddi enfeksiyon oran�, 100 hasta y�l� ba��na 1,7 olayd�r. Ayn� oran; g�nde iki kez 10 mg tofasitinib monoterapisi grubunda, 100 hasta y�l� ba��na 1,6 olay iken, plasebo grubunda 100 hasta y�l� ba��na 0 olay olarak ger�ekle�mi�tir. MTX grubunda ise bu oran 100 hasta y�l� ba��na 1,9 olay olmu�tur.
6, 12 veya 24 ay s�reli �al��malarda, g�nde iki kez 5 mg ve 10 mg tofasitinib art� DMARD gruplar�ndaki ciddi enfeksiyon oranlar� 100 hasta y�l� ba��na s�ras�yla 3,6 ve 3,4 olay iken, plasebo ile birlikte DMARD uygulanan grupta 100 hasta y�l� ba��na 1,7 olay olarak ger�ekle�mi�tir.
�laca maruz kalan t�m pop�lasyonlardaki uzun d�nem g�venlilik verilerinde, ciddi enfeksiyonlar�n genel oranlar� g�nde iki kez 5 mg ve g�nde iki kez 10 mg tofasitinib tedavisi alan gruplar i�in 100 hasta y�l� ba��na s�ras�yla 2,4 ve 3,0 olay olmu�tur. En yayg�n g�r�len ciddi enfeksiyonlar aras�nda, pn�moni, herpes zoster, idrar yolu enfeksiyonu, sel�lit, gastroenterit ve divertik�lit yer alm��t�r. F�rsat�� enfeksiyonlar da bildirilmi�tir (bkz. b�l�m 4.4).
En az bir ek kardiyovask�ler (KV) risk fakt�r� g�r�len, 50 ya��nda veya daha b�y�k romatoid artrit hastalar�yla yap�lan geni� (N=4.362), randomize, onay sonras� g�venlilik �al��mas�nda, TNF inhibit�rlerine k�yasla tofasitinib ile tedavi edilen hastalarda doza ba��ml� olarak artan insidansta ciddi enfeksiyonlar g�zlenmi�tir (bkz. b�l�m 4.4).
G�nde iki kez 5 mg tofasitinib, g�nde iki kez 10 mg tofasitinib ve TNF inhibit�rleri i�in ciddi enfeksiyon insidans oranlar� (%95 GA) her 100 hasta-y�l i�in s�ras�yla 2,86 (2,41-
3,37), 3,64 (3,11-4,23) ve 2,44 (2,02-2,92)'dur. TNF inhibit�rleri ile kar��la�t�r�ld���nda ciddi enfeksiyonlar�n risk oran� (HR), g�nde iki kez 10 mg tofatinib ve g�nde iki kez 5 mg tofasitinib i�in s�ras�yla 1,17 (0,92-1,50) ve 1,48 (1,17-1,87)'dur.
Ankilozan spondilit
Birle�tirilmi� faz 2 ve faz 3 randomize kontroll� klinik �al��malarda 48 haftaya kadar g�nde 2 kere tofasitinib 5 mg alan 316 hastada sadece 1 tane ciddi enfeksiyon g�r�lm��t�r (aseptik menenjit). Bu say� oranland���nda 100 hasta y�l� ba��na 0,43 olay denk gelmektedir.
Ya�l� hastalarda ciddi enfeksiyonlar
�al��ma I ila VI'ya dahil edilmi� olan 4,271 RA hastas�n�n 608'i 65 ya� ve �zerinde, bu hastalar�n da 85'i 75 ve �zeri ya�tad�r. Tofasitinib ile tedavi edilen 65 ya� ve �zeri hastalarda g�zlenen ciddi enfeksiyon s�kl���, 65 ya� alt� hastalarda g�r�lenden daha y�ksek olmu�tur (s�ras�yla 100 hasta y�l� ba��na 4,8 olaya kar�� 100 hasta y�l� ba��na 2,4 olay).
En az bir ek kardiyovask�ler (KV) risk fakt�r� g�r�len, 50 ya��nda veya daha b�y�k romatoid artrit hastalar�yla yap�lan geni� (N=4.362), randomize, onay sonras� g�venlilik �al��mas�nda, TNF inhibit�rlerine ve g�nde iki kere 5 mg tofasitinib kullan�m�na k�yasla g�nde iki kere 10 mg tofasitinib ile tedavi edilen 65 ya� ve �st� hastalarda ciddi enfeksiyonlarda art�� g�zlenmi�tir (bkz. b�l�m 4.4).
G�nde iki kez 5 mg tofasitinib, g�nde iki kez 10 mg tofasitinib ve TNF inhibit�rleri i�in 65 ya� ve �st� hastalarda ciddi enfeksiyon insidans oranlar� (%95 GA) her 100 hasta-y�l i�in s�ras�yla 4,03 (3,02-5,27), 5,85 (4,64-7,30) ve 3,73 (2,81-4,85)'dur.
TNF inhibit�rleri ile kar��la�t�r�ld���nda 65 ya� ve �st� hastalarda ciddi enfeksiyonlar�n risk oran� (HR), g�nde iki kez 5 mg tofatinib ve g�nde iki kez 10 mg tofasitinib i�in s�ras�yla 1,08 (0,74-1,58) ve 1,55 (1,10-2,19)'dur.
Giri�imsel olmayan onay sonras� g�venlilik �al��mas�ndan kaynaklanan ciddi enfeksiyonlar
Bir kay�ttan (US Corrona) RA hastalar�nda tofasitinibi de�erlendiren onay sonras� giri�imsel olmayan bir g�venlik �al��mas�ndan elde edilen veriler, g�nde bir kez uygulanan 11 mg tofasitinib i�in g�nde iki kez uygulanan 5 mg tofasitinibe k�yasla say�sal olarak daha y�ksek ciddi enfeksiyon insidans� g�zlemlendi�ini g�stermi�tir.
Tedavinin ba�lang�c�ndan 12 ay sonra her bir form�lasyonun uygunlu�undan elde edilen ham insidans oranlar� (%95 GA) (�rne�in ya� ve cinsiyete g�re ayarlanmam��) g�nde bir kez 11 mg tofasitinib ve g�nde iki kez 5 mg tofasitinib gruplar�nda s�ras�yla 100 hasta y�l� ba��na 3,45 (1,93, 5,69) ve 2,78 (1,74, 4,21) ve 36. ayda 4,71 (3,08, 6,91) ve 2,79 (2,01,
3,77) olayd�r. G�nde iki kez 5 mg tofasitinibe k�yasla g�nde 1 kez 11 mg tofasitinib i�in ayarlanmam�� risk oran� 12. ayda 1,30 (%95GA:0,67, 2,50) ve 36. ayda 1,93 (%95GA: 1,15, 3,24)'t�r. Veriler, nispeten b�y�k g�ven aral�klar� ve s�n�rl� takip s�resi ile g�zlemlenen olaylara sahip az say�da hastaya dayanmaktad�r.
Viral reaktivasyon
Tofasitinib ile tedavi edilen Japon ve Koreli hastalarda veya daha �nce iki veya daha fazla biyolojik DMARD alm�� olan uzun s�reli RA hastalar�nda, ALC de�eri 1,000 h�cre/mm'ten az olan hastalarda ve g�nde iki kere 10 mg doz alan hastalarda herpes zoster riski daha y�ksek olabilir (bkz. b�l�m 4.4).
En az bir ek kardiyovask�ler (KV) risk fakt�r� g�r�len, 50 ya��nda veya daha b�y�k romatoid artrit hastalar�yla yap�lan geni� (N=4.362), randomize, onay sonras� g�venlilik �al��mas�nda, TNF inhibit�rlerine ve tofasitinib ile tedavi edilen hastalarda herpes zoster olaylar�nda art�� g�zlenmi�tir (bkz. b�l�m 4.4). G�nde iki kez 5 mg tofasitinib, g�nde iki kez 10 mg tofasitinib ve TNF inhibit�rleri i�in herpes zoster insidans oranlar� (%95 GA) her 100 hasta-y�l i�in s�ras�yla 3,75 (3,22-4,34), 3,94 (3,38-4,57) ve 1,18 (0,90-1,52)'dur.
Laboratuvar testleri Lenfositler
Kontroll� RA klinik �al��malar�nda, g�nde iki kez hem 5 mg hem de 10 mg tofasitinib gruplar�nda yer alan hastalar�n %0,3'�nde ALC de�erleri 500 h�cre/mm seviyesinin alt�na d��m�� ve %1,9 hastada ise ALC de�eri 500-750 h�cre/mm aras�nda ��km��t�r.
Uzun d�nem RA g�venlilik pop�lasyonunda, g�nde iki kez hem 5 mg hem de 10 mg tofasitinib gruplar�nda yer alan hastalar�n %1,3'�nde ALC de�erleri 500 h�cre/mm seviyesinin alt�na d��m�� ve %8,4 hastada ise ALC de�eri 500-750 h�cre/mm aras�nda ��km��t�r.
ALC de�erinin 750 h�cre/mm'�n alt�na inmesi, ciddi enfeksiyonlar�n art��� ile ili�kili bulunmu�tur (bkz. b�l�m 4.4).
N�trofiller
Kontroll� RA klinik �al��malarda, g�nde iki kez hem 5 mg hem de 10 mg tofasitinib gruplar�nda yer alan hastalar�n %0,08'inde ANC de�erleri, 1,000 h�cre/mm seviyesinin alt�na d��m��t�r. Herhangi bir tedavi grubunda ANC de�eri 500 h�cre/mm de�erinin alt�na d��memi�tir. N�tropeni ve ciddi enfeksiyonlar�n olu�umu aras�nda net bir ili�ki izlenmemi�tir.
Uzun d�nem RA g�venlilik pop�lasyonunda, ANC'deki do�rulanm�� d���� insidans�, kontroll� klinik �al��malarda g�r�lenle tutarl� bir �ekilde seyretmi�tir (bkz. b�l�m 4.4).
Trombositler
Faz 3 kontroll� klinik �al��malarda kay�t ad�na uygun olmak i�in (RA, PsA, AS) hastalar�n trombosit d�zeyleri ≥100,000 h�cre/mm olmal�d�r. Bu nedenle, tofasitinib ile tedaviye ba�lamadan �nce trombosit d�zeyleri <100,000 h�cre/mm olan hastalar i�in herhangi bir bilgi mevcut de�ildir.
Karaci�er enzimi testleri
RA hastalar�nda, karaci�er enzimlerinde normalin �st limitinin (ULN) �� kat�ndan daha fazla art�� (3xULN) nadiren g�zlenmi�tir. Karaci�er enzimi art��� g�r�len bu hastalarda, e�lik eden DMARD dozunun azalt�lmas�, tofasitinib tedavisine ara verilmesi ya da tofasitinib dozunun azalt�lmas� gibi tedavi modifikasyonlar�, karaci�er enzimi seviyelerinin d����� ya da normal hale gelmesi ile sonu�lanm��t�r.
RA Faz 3 monoterapi �al��mas�n�n kontroll� b�l�m�nde (0-3 ay) (�al��ma I, bkz. b�l�m 5.1), plasebo, tofasitinib g�nde iki kez 5 mg ve g�nde iki kez 10 mg alan hastalar�n s�ras�yla
%1,65, %0,41 ve %0'�nda >3xULN ALT art��� g�zlenmi�tir. Bu �al��mada, plasebo,
tofasitinib g�nde iki kez 5 mg ve g�nde iki kez 10 mg alan hastalar�n s�ras�yla %1,65,
%0,41 ve %0'�nda >3x ULN AST art��� g�zlenmi�tir.
RA Faz 3 monoterapi �al��mas�nda (0-24 ay) (VI no.lu �al��ma, (bkz. b�l�m 5.1), MTX, tofasitinib g�nde iki kez 5 mg ve 10 mg alan hastalar�n s�ras�yla %7,1, %3,0 ve %3,0'�nde
>3xULN ALT art��� g�zlenmi�tir. Bu �al��mada, MTX, tofasitinib g�nde iki kez 5 mg ve g�nde iki kez 10 mg alan hastalar�n s�ras�yla %3,3, %1,6 ve %1,5'inde >3xULN AST art��� g�zlenmi�tir.
E� zamanl� DMARD alan hastalar�n dahil edildi�i RA Faz 3 �al��malar�n�n kontroll� b�l�m�nde (0-3 ay) (�al��ma II-V, bkz. b�l�m 5.1) plasebo, tofasitinib g�nde iki kez 5 mg ve g�nde iki kez 10 mg alan hastalar�n s�ras�yla %0,9, %1,24 ve %1,14'�nde >3xULN ALT art��� g�zlenmi�tir. Bu �al��malarda plasebo, tofasitinib g�nde iki kez 5 mg ve g�nde iki kez 10 mg alan hastalar�n s�ras�yla %0,72, %0,5 ve %0,31'inde >3x ULN AST art��� g�zlenmi�tir.
Monoterapi ile yap�lan uzun d�nem RA uzatma �al��malar�nda, tofasitinib g�nde iki kez 5 mg ve g�nde iki kez 10 mg alan hastalar�n s�ras�yla %1,1 ve %1,4'�nde >3xULN ALT art��� g�zlenmi�tir. Tofasitinib g�nde iki kez 5 mg ve g�nde iki kez 10 mg gruplar�n�n her ikisinde de <%1,0 oran�nda >3xULN AST art��� g�zlenmi�tir.
E� zamanl� DMARD alan hastalar�n dahil edildi�i uzun d�nem RA uzatma �al��malar�nda,
tofasitinib g�nde iki kez 5 mg ve g�nde iki kez 10 mg alan hastalar�n s�ras�yla %1,8 ve
%1,6's�nda >3xULN ALT art��� g�zlenmi�tir. Tofasitinib g�nde iki kez 5 mg ve g�nde iki kez 10 mg gruplar�n�n ikisinde de <%1,0 oran�nda >3xULN AST art��� g�zlenmi�tir.
En az bir ek kardiyovask�ler (KV) risk fakt�r� g�r�len, 50 ya��nda veya daha b�y�k romatoid artrit hastalar�yla yap�lan geni� (N=4.362), randomize, onay sonras� g�venlilik �al��mas�nda, ≥3xULN ALT y�kseli�i g�nde iki kez tofasitinib 5 mg, g�nde iki kez tofasitinib 10 mg ve TNF inhibit�rleri alan hastalarda s�ras�yla %6,01, %6,54 ve %3,77 hastada g�r�lm��t�r. ≥3xULN AST y�kseli�i g�nde iki kez tofasitinib 5 mg, g�nde iki kez tofasitinib 10 mg ve TNF inhibit�rleri alan hastalarda s�ras�yla %3,21, %4,57 ve %2,38 hastada g�r�lm��t�r.
Lipidler
Lipid parametrelerindeki art��lar (toplam kolesterol, LDL kolesterol, HDL kolesterol, trigliseritler) ilk olarak kontroll� �ift-k�r RA klinik �al��malar�nda tofasitinib ba�lanmas�ndan sonraki birinci ayda de�erlendirilmi�tir. Bu zaman noktas�nda g�zlenen art��lar ard�ndan stabil seyretmi�tir.
Kontroll� RA klinik �al��malar�nda �al��man�n ba�lang�c�ndan sonuna kadar (6-24 ay) olan s�redeki lipid parametrelerinde ortaya ��kan de�i�iklikler a�a��da �zetlenmi�tir:
Ortalama LDL kolesterol de�eri 12.ayda tofasitinib g�nde iki kez 5 mg kolunda %15 ve tofasitinib g�nde iki kez 10 mg kolunda %20 ve 24. ayda tofasitinib g�nde iki kez 5 mg kolunda %16 ve tofasitinib g�nde iki kez 10 mg kolunda %19 artm��t�r.
Ortalama HDL kolesterol de�eri 12.ayda tofasitinib g�nde iki kez 5 mg kolunda %17 ve tofasitinib g�nde iki kez 10 mg kolunda %18 ve 24. ayda tofasitinib g�nde iki kez 5 mg kolunda %19 ve tofasitinib g�nde iki kez 10 mg kolunda %20 artm��t�r.
Tofasitinib tedavisinin b�rak�lmas�n� takiben lipid seviyeleri ba�lang�� d�zeyine d�nm��t�r.
Tofasitinib ile tedavi edilen hastalarda ortalama LDL/HDL kolesterol oranlar� ve Apolipoprotein B (ApoB)/ApoAoranlar� esasen de�i�memi�tir.
Kontroll� bir RA klinik �al��mas�nda, LDL kolesterol ve Apo B seviyelerindeki art��lar, statin tedavisi ile tedavi �ncesi seviyelere d��m��t�r.
Uzun d�nem RA g�venlilik pop�lasyonunda, lipid parametrelerdeki art��lar, kontroll� klinik �al��malarda g�zlenenler ile tutarl� bir �ekilde seyretmi�tir.
En az bir ek kardiyovask�ler (KV) risk fakt�r� g�r�len, 50 ya��nda veya daha b�y�k romatoid artrit hastalar�yla yap�lan geni� (N=4.362), randomize, onay sonras� g�venlilik �al��mas�nda 24 ay boyunca lipid parametrelerinde de�i�iklikler a�a��da �zetlenmi�tir.
Ortalama LDL kolesterol de�erinde art�� 12.ayda tofasitinib g�nde iki kez 5 mg
tofasitinib, g�nde iki kez 10 mg tofasitinib ve TNF inhibit�r�nde s�ras�yla %13,80,
%17,04 ve %5,50 hastada g�r�lm��t�r. 24. ayda art�� s�ras�yla %12,71, %18,14 ve
%3,64 oran�ndad�r.
Ortalama HDL kolesterol de�erinde art�� 12.ayda tofasitinib g�nde iki kez 5 mg
tofasitinib, g�nde iki kez 10 mg tofasitinib ve TNF inhibit�r�nde s�ras�yla %11,71,
%13,63 ve %2,82 hastada g�r�lm��t�r. 24. ayda art�� s�ras�yla %11,58, %13,54 ve
%1,42 oran�ndad�r.
Miyokardiyal enfarkt�s�
Romatoid artrit
50 ya� ve �st� ve en az bir ek kardiyovask�ler risk fakt�r�ne sahip RA'l� hastalarda yap�lan geni� (N=4,362) randomize ruhsat onay� sonras� g�venlilik �al��mas�nda, g�nde iki kez 5
mg tofasitinib, g�nde iki kez 10 mg tofasitinib ve TNF inhibit�rleri i�in �l�mc�l olmayan miyokardiyal enfarkt�s insidans oranlar� (%95 GA) her 100 hasta y�l� ba��na s�ras�yla 0,37 (0,22, 0,57), 0,33 (0,19, 0,53) ve 0,16 (0,07, 0,31) olayl� hastad�r. TNF inhibit�rlerine k�yasla tofasitinib ile tedavi edilen hastalarda az say�da �l�mc�l miyokardiyal enfarkt�s� benzer oranlarda raporlanm��t�r (bkz. b�l�m 4.4 ve 5.1). Bu �al��ma, en az 1500 hastan�n 3 y�l boyunca takip edilmesini gerektirmi�tir.
NMSC harici maligniteler
Romatoid artrit
50 ya� ve �st� ve en az bir ek kardiyovask�ler risk fakt�r�ne sahip RA'l� hastalarda yap�lan geni� (N=4,362) randomize ruhsat onay� sonras� g�venlilik �al��mas�nda, g�nde iki kez 5 mg tofasitinib, g�nde iki kez 10 mg tofasitinib ve TNF inhibit�rleri i�in akci�er kanseri insidans oranlar� (%95 GA) her 100 hasta y�l� ba��na s�ras�yla 0,23 (0,12, 0,40), 0,32 (0,18,
0,51) ve 0,13 (0,05, 0,26) olayl� hastad�r (bkz. b�l�m 4.4 ve 5.1). Bu �al��ma, en az 1500 hastan�n 3 y�l boyunca takip edilmesini gerektirmi�tir.
G�nde iki kez 5 mg tofasitinib, g�nde iki kez 10 mg tofasitinib ve TNF inhibit�rleri i�in lenfoma insidans oranlar� (%95 GA) her 100 hasta y�l� i�in s�ras�yla 0,07 (0,02, 0,18), 0,11
(0,04, 0,24) ve 0,02 (0,00, 0,10) olayl� hastad�r (bkz. b�l�m 4.4 ve 5.1).
��pheli advers reaksiyonlar�n raporlanmas�
Ruhsatland�rma sonras� ��pheli ila� advers reaksiyonlar�n�n raporlanmas� b�y�k �nem ta��maktad�r. Raporlama yap�lmas�, ilac�n yarar / risk dengesinin s�rekli olarak izlenmesine olanak sa�lar. Sa�l�k mesle�i mensuplar�n�n herhangi bir ��pheli advers reaksiyonu T�rkiye Farmakovijilans Merkezi (TUFAM)'ne bildirmeleri gerekmektedir (www.titck.gov.tr; e- posta: tufam@titck.gov.tr; tel: 0 800 314 00 08; faks: 0 312 218 35
99)
4.9. Doz a��m� ve tedavisi
XELJANZ XR ile doz a��m� durumunda kullan�labilecek olan spesifik bir antidot bulunmamaktad�r. Doz a��m� durumunda hastan�n advers reaksiyonlara ili�kin belirti ve bulgular bak�m�ndan izlenmesi �nerilir.
Sa�l�kl� g�n�ll�lerde 100 mg'l�k tek bir doza kadar olan farmakokinetik veriler, uygulanan dozun %95'inden fazlas�n�n 24 saat i�inde elimine edilmesinin beklendi�ini g�stermektedir.
5. FARMAKOLOJ�K �ZELL�KLER
5.1. Farmakodinamik �zellikler
Farmakoterap�tik grup: Antineoplastik ve �mm�nomod�lat�r Ajanlar, Selektif Imm�nos�presanlar
ATC kodu: L04AA29
Etki mekanizmas�
Tofasitinib, JAK ailesinin g��l�, selektif bir inhibit�r�d�r. Enzim tayinlerinde tofasitinib, JAK1, JAK2, JAK3 ve daha d���k oranda olmak �zere TyK2'yi inhibe eder. Buna kar��l�k, tofasitinib, insan genomundaki di�er kinazlara kar�� y�ksek derecede se�icili�e sahiptir. Tofasitinib, insan h�crelerinde, tercihen fonksiyonel se�icilik g�stererek JAK2 �iftleri olarak sinyal ileten sitokin resept�rleri yerine JAK3 ve/veya JAK1 ile ili�kili heterodimerik sitokin resept�rleri ile sinyal iletimini inhibe eder. Tofasitinib ile JAK1 ve JAK3'�n inhibisyonu, interl�kinlerin (IL-2, -4, -7,- 9, -15 ve -21) ve tip I ve tip II interferonlar�n iletti�i sinyalleri azalt�r ve bu da imm�n ve inflamatuvar yan�t�n mod�lasyonu ile sonu�lan�r.
Farmakodinamik etkiler
RA hastalar�nda 6 aya kadar olan tofasitinib tedavisi, dola��mdaki CD16/56+ do�al �ld�r�c� (NK) h�cre say�s�nda doza ba�l� d����ler ile ili�kilidir, tahmini maksimum d����ler tedavi ba�lang�c�ndan yakla��k 8-10 hafta sonra ger�ekle�ir. Bu de�i�iklikler genellikle tedavinin sonland�r�lmas�ndan 2-6 hafta sonra ortadan kalkar. Tofasitinib ile tedavi, B h�cre say�lar�nda doza ba�l� art�� ile ili�kilidir. Dola��mdaki T-lenfosit say�lar� ve T-lenfosit alt tiplerindeki (CD3+, CD4+, CD8+) de�i�iklikler az miktarda ve tutars�z karakterdedir.
Uzun s�reli tedaviyi (tofasitinib tedavisinin ortalama s�resi yakla��k 5 y�l) takiben CD4+ ve CD8+ say�lar�nda ortalama azalma ba�lang��tan itibaren s�ras�yla %28 ve %27 oran�nda g�zlenmi�tir. K�sa s�reli dozlamay� takiben g�zlemlenen d�����n aksine, CD16/56+ do�al �ld�r�c� h�cre say�lar�nda ba�lang��tan itibaren ortalama %73'l�k bir art�� g�zlenmi�tir. CD19+B h�crelerinin say�s�nda uzun s�reli tofasitinib tedavisinden sonra ba�ka art��lar g�zlenmemi�tir. Lenfosit alt tiplerindeki t�m de�i�iklikler, tedavinin ge�ici olarak kesilmesini takiben ba�lang�� de�erine do�ru geri d�nm��t�r. Ciddi veya f�rsat�� enfeksiyonlar veya herpes zoster ve lenfosit alt tiplerinin say�s� aras�nda ili�ki oldu�una dair bir kan�t yoktur (mutlak lenfosit say�m� takibi i�in bkz. b�l�m 4.2).
RA hastalar�na 6 ayl�k tofasitinib dozu sonras� toplam serum IgG, IgM ve IgA seviyelerindeki de�i�iklikler k���kt�r, doz ba��ml� de�ildir ve plaseboda g�r�lenlerle benzerdir ki bu da sistemik humoral supresyonun yoklu�una i�aret etmi�tir.
RA hastalar�nda tofasitinib ile tedaviyi takiben, serum C-reaktif proteini seviyelerinde (CRP) h�zl� bir d���� g�zlenmi� ve bu d���� tedavi boyunca s�rd�r�lm��t�r. Tofasitinib tedavisi ile g�zlenen CRP de�i�iklikleri, tedavi durdurulduktan sonraki 2 hafta i�inde tam olarak eski haline d�nmemi�tir, bu da ilac�n yar� �mr� ile kar��la�t�r�ld���nda daha uzun bir farmakodinamik aktivite s�resini i�aret etmektedir.
A�� �al��malar�
G�nde iki kez 10 mg tofasitinib veya plasebo ile tedavi ba�lat�lan RA hastalar� �zerinde ger�ekle�tirilen kontroll� bir klinik �al��mada, influenza a��s�na yan�t verenlerin say�s� her iki grupta benzerdir: Tofasitinib (%57) ve plasebo (%62). Pn�mokokal polisakkarit a��s�na yan�t verenlerin say�s� ��yledir: hem tofasitinib hem de MTX alan hastalarda %32;
tofasitinib monoterapisi alan hastalarda %62; MTX monoterapisi alan hastalarda %62 ve plasebo alan hastalarda %77'dir. Bunun klinik a��dan anlam� bilinmemekle beraber, uzun s�re boyunca g�nde iki kez 10 mg tofasitinib alan hastalarda influenza ve pn�mokokal polisakkarit a��lar� ile yap�lan ayr� bir a�� �al��mas�nda da yine benzer sonu�lar al�nm��t�r.
RA hastalar�nda, g�nde iki kez 5 mg tofasitinib veya plasebo ile 12 haftal�k tedavi ba�lat�lmadan 2 ila 3 hafta �ncesinde canl� aten�e bir herpes vir�s a��s� ile imm�nize edilmi� daha �nce MTX kullanan hastalar �zerinde kontroll� bir �al��ma yap�lm��t�r. 6. haftada hem tofasitinib ve hem de plasebo ile tedavi edilen hastalarda VZV'ye humoral ve h�cresel yan�tlara dair kan�tlar g�r�lm��t�r. Bu yan�tlar 50 ya� ve �zeri sa�l�kl� g�n�ll�lerde g�zlemlenmi� olanlara benzerdir. Ge�mi�te varisella enfeksiyonu �yk�s� ve ba�lang�� seviyesinde anti-varisella antikoru bulunmayan bir hastada a��lamadan 16 g�n sonra varisellan�n a�� su�unun yay�l�m� g�zlenmi�tir. Tofasitinib tedavisi kesilmi� ve hasta standart antiviral ila� tedavisi sonras� iyile�mi�tir. Bu hasta daha sonra a��ya gecikmi� olsa da sa�lam, h�moral ve h�cresel bir yan�t vermi�tir (bkz. b�l�m 4.4).
Klinik etkililik ve g�venlilik
Romatoid artrit
Tofasitinib film kapl� tabletin etkililik ve g�venlili�i, 18 ya��ndan b�y�k ve Amerikan Romatoloji Derne�inin kriterlerine g�re aktif RA tan�s� konmu� hastalar�n dahil oldu�u 6 randomize, �ift-k�r, �ok merkezli klinik �al��mada de�erlendirilmi�tir. Tablo 2'te �al��ma tasar�m�na ve pop�lasyonun �zelliklerine dair bilgiler bulunmaktad�r.
Tablo 2: RA'l� hastalarda g�nde iki defa 5 mg ve 10 mg tofasitinib dozlar�n�n faz 3 klinik �al��malar�
�al��malar | �al��ma I (ORAL Solo) | �al��ma II (ORAL Sync) | �al��ma III (ORAL Standard) | �al��ma IV (ORAL Scan) | �al��ma V (ORAL Step) | �al��ma VI (ORAL Start) | �al��ma VII (ORAL Strategy) |
Pop�lasyon | DMARD- IR | DMARD- IR | MTX-IR | MTX-IR | TNFi-IR | MTX-naifa | MTX-IR |
Kontrol | Plasebo | Plasebo | Plasebo | Plasebo | Plasebo | MTX | MTX, ADA |
�nceki tedavi |
Yok |
csDMARD 'lar |
MTX |
MTX |
MTX |
Yok | 3 Paralel kollu: monoterapi +MTX |
Tofasitinib
Tofasitinib
ADA+MTX
�nemli �zellikler | Monoterapi | �e�itli csDMARD 'lar | Aktif kontrol (ADA) | X-Ray | TNFi-IR | Monoterapi , Aktif komperat�r (MTX), X-Ray | MTX'li ve MTX'siz tofasitinibin MTX'li ADA ile kar��la�t�r�lma |
Tedavi alan hasta say�s�) | 610 | 792 | 717 | 797 | 399 | 956 | 1,146 |
Toplam �al��ma | 6 ay | 1 y�l | 1 y�l | 2 y�l | 6 ay | 2 y�l | 1 y�l |
Koprimer etkililik sonlan�m noktas�c | 3. ay: ACR20 HAQ-DI DAS28- 4(ESR)<2,6 | 6. ay: ACR20 DAS28- 4 (ESR)<2,6 3. ay: HAQ-DI | 6. ay: ACR20 DAS28- 4 (ESR)<2,6 3. ay: HAQ-DI | 6. ay: ACR20 mTSS DAS28- 4 (ESR)<2,6 3. ay: HAQ-DI | 3. ay: ACR20 HAQ-DI DAS28- 4 (ESR)<2,6 | 6. ay: mTS S ACR 70 | 6. ay: ACR50 |
G�nde 2 kez 5 mg veya 10 mg tofasitinib i�in zorunlu plasebo kurtarma zaman� |
3. ay |
6. ay (3. ay'da tofasinitibe k�yasla �i� ve hassas eklem say�s�nda <%20 geli�me g�r�nen plasebo denekleri) |
3. ay |
NA |
NA | ||
5.2. Farmakokinetik �zellikler
Genel �zelliklerTofasitinibin oral yoldan 11 mg uzat�lm�� sal�ml� tablet uygulamas�n� takiben pik plazma konsantasyonuna 4 saatte ula�m��t�r ve yar�lanma �mr� yakla��k 6 saat olmu�tur. G�nde bir kere al�m�n� takiben ihmal edilebilir d�zeydeki ak�m�lasyon ile kararl� durum konsantasyonuna 48 saat i�inde ula��lm��t�r. Kararl� durum tofasitinib EAA ve Cmaks de�erleri g�nde iki kere 5 mg tofasitinib film tablet ve g�nde bir kere 11 mg uzat�lm�� sal�ml� film kapl� tablette ayn�d�r.
Emilim:
Tofasitinib 11 mg uzat�lm�� sal�ml� film kapl� tabletlerin y�ksek ya� i�erikli bir yemek ile birlikte al�nmas�, EAA'da herhangi bir de�i�ikli�e neden olmazken, C'� %27 oran�nda azaltm��t�r.
Da��l�m:
�ntraven�z uygulamadan sonra da��l�m hacmi 87 L'dir. Kanda dola�an tofasitinibin yakla��k %40'� plazma proteinlerine ba�lan�r. Tofasitinib, a��rl�kl� olarak alb�mine ba�lanmaktad�r ve α1-asit glikoproteine ba�lan�yor gibi g�r�nmemektedir. Tofasitinib, alyuvarlar ve plazma aras�nda e�it olarak da��lmaktad�r.
Biyotransformasyon:
Tofasitinib i�in klirens mekanizmalar� yakla��k %70 hepatik metabolizma ve ana ilac�n
%30'unun b�brek yoluyla at�lmas� �eklindedir. Tofasitinib, esas olarak CYP3A4 arac�l���yla metabolize edilir; CYP2C19'un min�r bir katk�s� vard�r. �nsanlar �zerinde yap�lan bir radyoaktif i�aretleme �al��mas�nda, dola��mdaki toplam radyoaktivitenin
%65'inden fazlas� de�i�memi� etkin maddeye, geri kalan %35'i, her biri %8'den az radyoaktiviteye kar��l�k gelen 8 metabolite atfedilmi�tir. T�m metabolitler hayvan t�rlerinde g�zlenmi�tir ve JAK1/3 inhibisyonu i�in tofasitinibden 10 kat daha d���k bir g�ce sahip olduklar� tahmin edilmektedir. �nsan �rneklerinde stereo d�n���m kan�tlar� tespit edilmemi�tir. Tofasitinibin farmakolojik aktivitesi ana molek�le atfedilmi�tir. In vitro ortamda tofasitinib MDR1 i�in bir substrat olmas�na kar��n meme kanseri diren� proteini (BCRP), OATP1B1/1B3 veya OCT1/2 i�in bir substrat inhibit�r de�ildir.
Eliminasyon:
Tofasitinib i�in klirens mekanizmalar� yakla��k %70 hepatik metabolizma ve ana ilac�n
%30'unun b�brek yoluyla at�lmas� �eklindedir.
Do�rusall�k /do�rusal olmayan durum:
Ge�erli de�ildir.
Hastalardaki karakteristik �zellikler
Hastalardaki farmakokinetik
CYP enzimlerinin enzimatik aktivitesi RA hastalar�nda kronik inflamasyon dolay�s�yla azalmaktad�r. RA hastalar�nda, tofasitinibin oral klirensi zamanla de�i�memektedir ki bu da tofasitinib ile tedavinin CYP enzim aktivitesini normalle�tirmedi�ine i�aret etmektedir.
RA hastalar� �zerinde yap�lan pop�lasyon PK analizi v�cut a��rl���n�n a��r� u�lar�nda (40 kg, 140 kg) tofasitinibin sistemik maruziyetinin (EAA) 70 kg bir hastan�nkine benzer (%5 dahilinde) oldu�unu g�stermi�tir. 80 ya��ndaki ya�l� hastalar�n EAA de�erinin 55 ya� �eklindeki ortalamaya g�re %5'ten d���k bir oranda daha az oldu�u tahmin edilmi�tir. Kad�nlar�n EAA de�erinin erkeklere oranla %7 daha d���k oldu�u �ng�r�lmektedir. Mevcut veriler, Beyaz, Siyah ve Asyal� hastalar aras�nda tofasitinib EAA de�eri a��s�ndan �nemli farklar bulunmad���n� g�stermi�tir. V�cut a��rl��� ve da��l�m hacmi aras�nda, daha hafif hastalarda daha y�ksek pik (C) ve daha d���k dip (C) konsantrasyonlar ile sonu�lanan, neredeyse lineer bir ili�ki g�zlenmi�tir. Ancak, bu fark klinik a��dan ilgili kabul edilmemi�tir. Tofasitinibin EAA de�eri a��s�ndan g�n�ll�ler aras�ndaki de�i�kenlik (y�zde varyasyon katsay�s�) yakla��k %27 olarak tahmin edilmektedir.
Aktif PsA'l� veya AS'li hastalarda PK analizi pop�lasyonundan elde edilen sonu�lar RA hastalar�ndan elde edilenler ile tutarl�d�r.
B�brek yetmezli�i
Hafif (kreatinin klirensi 50-80 mL/dak), orta (kreatinin klirensi 30-49 mL/dak) ve �iddetli (kreatinin klirensi <30 mL/dak) b�brek yetmezli�i olan bireylerde EAA de�erinde, sa�l�kl� bireylerle kar��la�t�r�ld���nda s�ras�yla %37, %43 ve %123 oran�nda art�� g�zlenmi�tir (bkz. b�l�m 4.2). Son evre b�brek yetmezli�i (SEBY) olan bireylerde, diyalizin tofasitinib toplam klirensine katk�s� g�receli olarak az olmu�tur. 10 mg'l�k tek bir dozu takiben, SEBY'li bireylerde diyaliz olmayan g�nlerde �l��len konsantrasyonlara dayal� ortalama EAA normal b�brek fonksiyonu olan bireylere k�yasla yakla��k %40 oran�nda daha y�ksektir (%90 g�ven aral�klar�: %1,5-95). Klinik �al��malarda tofasitinib ba�lang�� kreatinin klirens seviyeleri (Cockroft-Gault denklemi ile tahmin edilen �ekilde) 40 mL/dakikadan az olan hastalarda de�erlendirilmemi�tir (bkz. b�l�m 4.2).
Karaci�er yetmezli�i
Hafif (Child Pugh A) ve orta derecede (Child Pugh B) karaci�er yetmezli�i olan bireylerde EAA de�erinde, normal karaci�er fonksiyonuna sahip olan bireylerle kar��la�t�r�ld���nda s�ras�yla %3 ve %65 oran�nda art�� g�zlenmi�tir. Klinik �al��malarda, tofasitinib �iddetli (Child Pugh C) karaci�er yetmezli�i olan hastalarda (bkz. b�l�m 4.2 ve 4.4), veya hepatit B ya da C i�in pozitif sonu� al�nan hastalarda de�erlendirilmemi�tir.
�la� etkile�imi
Tofasitinib CYP'ler i�in (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 ve
CYP3A4) bir inhibit�r veya ind�kleyici UGT'ler i�in (UGT1A1, UGT1A4, UGT1A6, UGT1A9 ve UGT2B7) bir inhibit�r de�ildir. Tofasitinib klinik olarak anlaml� konsantrasyonlarda MDR1, OATP1B1/1B3, OCT2, OAT1/3 veya MRP i�in bir inhibit�r de�ildir.
Uzat�lm�� sal�ml� tablet ile film kapl� tabletilerin FK kar��la�t�rmas�
G�nde bir kere tofasitinib 11 mg uzat�lm�� sal�ml� film kapl� tablet g�nde 2 kere tofasitinib 5 mg film kapl� tablet ile e�de�er FK (EAA ve C) g�stermi�tir.
5.3. Klinik �ncesi g�venlilik verileri
Klinik d��� �al��malarda, ba����kl�k ve hematopoietik sistemlerde tofasitinibin farmakolojik �zellikleri (JAK inhibisyonu) ile ili�kilendirilen etkiler g�zlenmi�tir. Klinik ile ili�kilendirilebilecek dozlarda, bakteriyel ve viral enfeksiyonlar ve lenfoma gibi imm�nos�presyondan kaynaklanan ikincil etkiler g�r�lm��t�r. Lenfoma 8 yeti�kin maymunun 3'�nde klinik tofasitinib maruziyet seviyesinin 6 veya 3 kat�nda (insanlarda g�nde iki kez 5 mg veya 10 mg dozunda ba�l� olmayan EAA) g�r�lm��t�r ve g�nde 2 kere 5 mg veya 10 mg dozlar�nda klinik maruziyet seviyesinin 5 veya 2,5 kat�nda 14 gen� maymunun hi�birinde g�r�lmemi�tir. Lenfomalar i�in Hi� Advers Etki G�zlenmeyen Seviyede (NOAEL) maymunlarda maruziyet g�nde iki kez 5 mg veya 10 mg yakla��k olarak klinik maruziyet seviyesinin 1 veya 0,5 kat�na e�ittir. �nsanlara uygulanan dozlar�n olduk�a �zerinde olan dozlar ile ortaya ��kan di�er bulgular aras�nda karaci�er ve gastrointestinal sistemler �zerindeki etkiler yer alm��t�r.
Gen mutasyonlar� ve krozomal aberasyonlara y�nelik yap�lan bir dizi in vitro ve in vivo
testlerin bulgular�na g�re, tofasitinib mutajenik veya genotoksik de�ildir.
Tofasitinibin karsinojenik potansiyeli 6 ayl�k rasH2 transjenik fare ve 2 y�l s�reli s��an karsinojenite �al��malar�nda de�erlendirilmi�tir. Tofasitinib g�nde iki kere 5 mg veya 10 mg dozlarda maruziyet seviyesinin 38 kat� veya 19 kat� kadar olan seviyelerde s��anlarda karsinojenik de�ildir. S��anlarda benign testik�ler interstisyel (Leydig) h�cre t�m�rleri g�zlenmi�tir; s��anlardaki benign Leydig h�cre t�m�rleri insanlarda Leydig h�cre t�m�rleri riski ile ba�lant�l� de�ildir. Hibernomalar (kahverengi ya� dokusunun malignitesi) g�nde iki kere 5 mg veya 10 mg dozlarda maruziyet seviyesinin 83 kat� veya klinik maruziyet seviyesinin 41 kat� seviyelerinde di�i s��anlarda g�zlenmi�tir. Di�i s��anlarda g�nde iki kere 5 mg veya g�nde iki kere 10 mg dozlarda klinik maruziyet seviyesinin 187 veya 94 kat� seviyelerinde benign timomalar g�r�lm��t�r.
Tofasitinibin s��anlar ve tav�anlarda teratojenik oldu�u ve s��anlarda di�i fertilitesini (azalan gebelik oran�; corpora lutea, implantasyon yeri ve canl� fet�s say�lar�nda d���� ve erken resorpsiyonlarda art��), part�risyonu ve peri/postnatal geli�imi etkiledi�i g�sterilmi�tir. Tofasitinibin erkek fertilitesi, sperm motilitesi veya sperm konsantrasyonu �zerinde bir etkisi yoktur. Tofasitinib, emziren s��anlar�n s�t�nde dozu takip eden 1 ila 8 saatte, serumdakilerin yakla��k 2 kat� konsantrasyonlarda at�lm��t�r. J�venil s��anlar ve tav�anlarda yap�lan �al��malarda onaylanm�� insan dozundaki maruziyetlerde erkek yada di�ilerde kemik geli�iminde tofasitinib ile illi�kili bir etki yoktur.
J�venil hayvan �al��malar�nda, pediyatrik pop�lasyonlar�n yeti�kinlere k�yasla daha y�ksek bir duyarl�l��a sahip oldu�unu g�steren, tofasitinib ile ili�kili hi�bir bulgu g�zlenmemi�tir. J�venil s��an fertilite �al��mas�nda, geli�imsel toksisiteye dair bir kan�t bulunmam��, cinsel olgunla�ma �zerinde hi�bir etki olmam�� ve cinsel olgunluktan sonra �reme toksisitesine (�iftle�me ve do�urganl�k) dair hi�bir kan�t kaydedilmemi�tir.
1 ayl�k j�venil s��an ve 39 haftal�k j�venil maymun �al��malar�nda, ba����kl�k ve hematoloji parametreleri �zerinde JAK1/3 ve JAK2 inhibisyonu ile uyumlu tofasitinib ile ili�kili etkiler g�zlenmi�tir. Bu etkiler tersine �evrilebilir olup benzer maruziyetlerde yeti�kin hayvanlarda da g�zlemlenenlerle tutarl�d�r.
6. FARMAS�T�K �ZELL�KLER
6.1. Yard�mc� maddelerin listesi
Tablet �ekirde�i Sorbitol (E420) Hidroksietil sel�loz Kopovidon Magnezyum stearat
Film kaplama
Sel�loz asetat
Hidroksipropil sel�loz (E463)
Renk kaplamas� (Opadry pembe 03K140024)
HPMC 2910 /Hipromelloz (E 464) Titanyum dioksit (E171)
Triasetin
K�rm�z� demir oksit (E172)
6.2. Ge�imsizlikler
Ge�erli de�ildir.
6.3. Raf �mr�
24 ay
6.4. Saklamaya y�nelik �zel tedbirler
25°C'nin alt�ndaki oda s�cakl���nda saklanmal�d�r. Nemden korumak i�in orijinal ambalaj�nda saklanmal�d�r.
6.5. Ambalaj�n niteli�i ve i�eri�i
28 film kapl� tablet i�eren folyo / folyo blisterleri.
6.6. Be�eri t�bbi �r�nden arta kalan maddelerin imhas� ve di�er �zel �nlemler
Kullan�lmam�� olan �r�nler ya da at�k materyaller ‘T�bbi At�klar�n Kontrol� Y�netmeli�i' ve ‘Ambalaj At�klar�n�n Kontrol� Y�netmelikleri'ne uygun olarak imha edilmelidir.
 Asperger Sendromu
Asperger sendromu, otistik gurubun bir b�l�m� olan bir �z�rd�r. Bu genelde,
gurubun daha ”y�ksek” taraf�nda yer ald��� d���n�len ki�ilere uygun bir tan�d�r.
Asperger Sendromu
Asperger sendromu, otistik gurubun bir b�l�m� olan bir �z�rd�r. Bu genelde,
gurubun daha ”y�ksek” taraf�nda yer ald��� d���n�len ki�ilere uygun bir tan�d�r. |
 Travma Sonras� Bunal�m�
Travmatik bir olay, g�nl�k ola�an olaylar�n d���nda olan ve ki�iyi derinden
rahats�z eden bir olayd�r.Bir�ok olay b�yle bir etki g�sterebilir.
Travma Sonras� Bunal�m�
Travmatik bir olay, g�nl�k ola�an olaylar�n d���nda olan ve ki�iyi derinden
rahats�z eden bir olayd�r.Bir�ok olay b�yle bir etki g�sterebilir. |
�LA� E�DE�ERLER�
| E�de�er �la� Ad� | Barkodu | �la� Fiyat� |
|---|---|---|
| TOFANZI | 8699511099218 | 7,389.67TL |
| TOFIK | 8680199002010 | 7,916.59TL |
| XELJANZ | 8681308090003 | 8,169.02TL |
| Di�er E�de�er �la�lar |
 |
Artrit Artrit, olduk�a yayg�n bir hastal�kt�r ancak iyi anla��lamam��t�r. Asl�nda �artrit� tek bir hastal���n ad� de�ildir; eklem a�r�s� veya eklem hastal�klar�n� adland�rman�n gayri resmi yoludur. |
 |
L�semi Kan Kanseri L�semi, kan kanseridir ve v�cudunun kan olu�turan dokular�n�n hastalanmas� anlam�na gelir. Bir�ok l�semi t�r� vard�r; baz� l�semi t�rleri �ocuklarda baz�lar� da yeti�kinlerde s�k g�r�l�r. |
|
Artrit Artrit, olduk�a yayg�n bir hastal�kt�r ancak iyi anla��lamam��t�r. Asl�nda �artrit� tek bir hastal���n ad� de�ildir; eklem a�r�s� veya eklem hastal�klar�n� adland�rman�n gayri resmi yoludur. |
�LA� GENEL B�LG�LER�
Pfizer �la�lar� Ltd.�ti.
| Sat�� Fiyat� | 11723.11 TL [ 26 Apr 2024 ] |
| �nceki Sat�� Fiyat� | 11723.11 TL [ 22 Apr 2024 ] |
| Original / Jenerik | Original �la� |
| Re�ete Durumu | Normal Re�eteli bir ila�d�r. |
| Barkodu | 8681308031044 |
| Etkin Madde | Tofacitinib |
| ATC Kodu | L04AA29 |
| Birim Miktar | 11 |
| Birim Cinsi | MG |
| Ambalaj Miktar� | 28 |
| Antineoplastik ve �mm�nomod�lat�r Ajanlar > �mm�nsupresif Ajanlar |
| Yerli ve Be�eri bir ila�d�r. |