SARCLISA 100 mg / 5 ml infüzyonluk çözelti hazırlamak için konsantre Kısa Ürün Bilgisi
{ Isatuksimab }
1. BEŞERİ TIBBİ ÜRÜNÜN ADI
SARCLISA 100 mg/5 ml infüzyonluk çözelti hazırlamak için konsantre Steril
2. KALİTATİF VE KANTİTATİF BİLEŞİM
Etkin madde
İnfüzyonluk çözelti hazırlamak için konsantrenin her bir mL'si 20 mg isatuximab içermektedir.
Her bir flakon 5 mL konsantre içinde 100 mg isatuximab içermektedir (100 mg/5 mL).
İsatuksimab, memeli hücre dizisinden (Çin Hamster Yumurtalık, CHO) üretilen bir immünoglobulin G1 (IgG1) monoklonal antikorudur (mAb).
Yardımcı maddeler
Yardımcı maddelerin tam listesi için 6.1'e bakınız.
3. FARMASÖTİK FORMU
İnfüzyonluk çözelti hazırlamak için konsantre
Renksiz ila açık sarı renkli, esas olarak görünür partikül içermez.
4. KLİNİK ÖZELLİKLER
4.1. Terapötik endikasyonlar
SARCLISA, lenalidomid ve bir proteazom inhibitörü dahil daha önce en az iki tedavi alan ve son tedavilerinde progresyon görülen, nüks ve dirençli erişkin multipl miyelom (MM) hastalarının tedavisinde, pomalidomid ve deksametazon ile kombine kullanımda endikedir.
4.2. Pozoloji ve uygulama şekli
SARCLISA resüsitasyon imkanı mevcut olan bir ortamda bir sağlık uzmanı tarafından uygulanmalıdır.
Premedikasyon
İnfüzyon reaksiyonlarının riski ve şiddetini azaltmak için, SARCLISA infüzyonundan önce aşağıdaki tıbbi ürünlerle premedikasyon kullanılmalıdır:
Deksametazon 40 mg, oral veya intravenöz (veya 75 yaş ve üzerindeki hastalar için 20 mg oral veya intravenöz).
Asetaminofen 650 mg ila 1000 mg, oral (veya eşdeğeri).
H2 antagonistleri (ranitidin 50 mg intravenöz veya eşdeğeri [örn. simetidin]) veya oral proton pompası inhibitörleri (örn. omeprazol, esomeprazol).
Difenhidramin 25 mg ila 50 mg, intravenöz veya oral (veya eşdeğeri [örn. setirizin, prometazin, deksklorfeniramin]). En az ilk 4 infüzyon için intravenöz yol tercih edilmektedir.
Yukarıda önerilen deksametazon dozu (oral veya intravenöz), infüzyondan önce sadece bir kere, premedikasyon ve omurga tedavi kapsamında, isatuximab ve pomalidomid uygulamasından önce uygulanacak toplam doza karşılık gelmektedir.
Önerilen premedikasyon ajanları, SARCLISA infüzyonuna başlamadan 15-60 dakika önce uygulanmalıdır. İlk dört SARCLISA uygulamasından sonra infüzyon reaksiyonu yaşamayan hastaların daha sonraki premedikasyon ihtiyaçları tekrar düşünülebilir.
Nötropeni yönetimi
Nötropeni riskini azaltmak için koloni uyarıcı faktörlerin (örn. G-CSF) kullanımı düşünülmelidir. Derece 4 nötropeni olayı durumunda SARCLISA uygulaması, nötrofil sayısı en az 1,0 × 10/L'ye yükselene kadar ertelenmelidir (bkz. Bölüm 4.4).
Pozoloji/uygulama sıklığı ve süresi:
SARCLISA'nın önerilen dozu, Tablo 1'deki dozlama planına göre, pomalidomid ve deksametazonla (isatuximab rejimi) kombinasyon halinde intravenöz infüzyon olarak 10 mg/kg vücut ağırlığıdır:
Tablo 1: Pomalidomid ve deksametazonla kombinasyon halinde SARCLISA dozlama planı
Sikluslar | Dozlama Planı |
1. Siklus | 1., 8., 15. ve 22. günler (haftalık) |
2. Siklus ve sonrası | 1., 15. günler (2 haftada bir) |
Her bir tedavi siklusu 28 günlük bir dönemden oluşmaktadır. Tedavi hastalık progresyonu veya kabul edilemez toksisite meydana gelene kadar tekrarlanmaktadır.
SARCLISA ile uygulanan diğer tıbbi ürünler için, ilgili güncel kısa ürün bilgilerine bakınız.
Uygulama programı dikkatle izlenmelidir. Planlanan bir SARCLISA dozu kaçırılırsa, doz en kısa zamanda uygulanmalı ve doz programı tedavi aralığı korunarak uygun şekilde ayarlanmalıdır.
Doz ayarlamaları
SARCLISA dozunun azaltılması önerilmemektedir.
Hastalar infüzyon reaksiyonu yaşarsa uygulama ayarlamaları yapılmalıdır (bkz. "Uygulama şekli").
SARCLISA ile birlikte uygulanan diğer tıbbi ürünler için, ilgili güncel kısa ürün bilgisi göz önünde bulundurulmalıdır.
Uygulama şekli:
SARCLISA intravenöz kullanım içindir. Tıbbi ürünün uygulanmadan önce seyreltilmesine ilişkin talimatlar için Bölüm 6.6'ya bakınız.
İnfüzyon hızları
Seyreltildikten sonra SARCLISA infüzyonu, intravenöz yoldan aşağıdaki Tablo 2'de sunulan infüzyon hızlarında uygulanmalıdır(bkz.Bölüm5.1).Yalnızca infüzyon reaksiyonu
Tablo 2: SARCLISA uygulamasının infüzyon hızları
| Seyreltme hacmi | İlk hız | İnfüzyon reaksiyonunun olmaması | Kademeli hız | Maksimum hız |
İlk infüzyon | 250 mL | 25 mL/saat | 60 dakika | 30 dakikada bir 25 mL/saat | 150 mL/saat |
İkinci infüzyon | 250 mL | 50 mL/saat | 30 dakika | 30 dakika boyunca 50 mL/saat ardından 30 dakikada bir 100 mL/saat artış | 200 mL/saat |
Sonraki infüzyonlar | 250 mL | 200 mL/saat |
|
| 200 mL/saat |
Hasta infüzyon reaksiyonu yaşarsa uygulamada ayarlamalar yapılmalıdır (bkz. Bölüm 4.4):
Derece 2 (orta) infüzyon reaksiyonu yaşayan hastalarda, infüzyona geçici olarak ara verilmesi düşünülmelidir ve ek semptomatik tıbbi ürünler uygulanabilir. Derece ≤ 1'e (hafif) iyileşmeden sonra, SARCLISA infüzyonuna ilk infüzyon hızının yarısında ve yakın izlem ve gerektikçe destekleyici bakımla devam edilebilir. 30 dakikadan sonra semptomlar tekrar etmezse, infüzyon hızı Tablo 2'de gösterildiği gibi ilk hıza geri çıkarılabilir ve daha sonra kademeli olarak yükseltilebilir.
SARCLISA infüzyonuna ara verilmesinden sonra semptomlar hızlı bir şekilde çözülmezse ve Derece ≤ 1'e iyileşmezse, uygun tıbbi ürünlerle ilk iyileşmeden sonra tekrar ederse veya hastaneye yatış gerektirirse ya da yaşamı tehdit ederse (Derece ≥ 3), SARCLISA tedavisi kalıcı olarak durdurulmalı ve gerektikçe ek destekleyici tedaviler uygulanmalıdır.
Özel popülasyonlara ilişkin ek bilgiler:
Geriyatrik popülasyon:
Popülasyon farmakokinetik analizine göre, yaşlı hastalarda önerilen bir doz ayarlaması bulunmamaktadır.
Böbrek yetmezliği:
Popülasyon farmakokinetik analizi ve klinik güvenliliğe göre, hafif ila şiddetli böbrek yetmezliği olan hastalarda önerilen bir doz ayarlaması bulunmamaktadır (bkz. Bölüm 5.2).
Karaciğer yetmezliği:
Popülasyon farmakokinetik analizine göre, hafif karaciğer yetmezliği olan hastalarda önerilen bir doz ayarlaması bulunmamaktadır. Orta dereceli ila şiddetli karaciğer yetmezliği olan hastalardaki veriler sınırlıdır (bkz. Bölüm 5.2), fakat bu hastalarda doz ayarlaması gerektiğine işaret eden bir kanıt bulunmamaktadır.
Pediyatrik popülasyon:
SARCLISA'nın 18 yaşından küçük çocuklardaki güvenliliği ve etkililiği kanıtlanmamıştır. Veri bulunmamaktadır.
4.3. Kontrendikasyonlar
Etkin madde
4.4. Özel kullanım uyarıları ve önlemleri
İnfüzyon reaksiyonları
SARCLISA ile tedavi edilen hastaların %38,2'sinde çoğu hafif veya orta dereceli infüzyon reaksiyonları gözlemlenmiştir (bkz. Bölüm 4.8). Tüm infüzyon reaksiyonları ilk SARCLISA infüzyonu sırasında başlamış ve infüzyonların %98'inde aynı gün içinde çözülmüştür. İnfüzyon reaksiyonlarının en yaygın semptomları arasında, dispne, öksürük, üşüme ve bulantı bulunmaktadır. En yaygın şiddetli belirtiler ve semptomlar arasında hipertansiyon ve dispne bulunmaktadır (bkz. Bölüm 4.8).
İnfüzyon reaksiyonlarının riskini ve şiddetini azaltmak için, SARCLISA infüzyonundan önce hastalara asetaminofen, H2 antagonistleri veya proton pompası inhibitörleri, difenhidramin veya eşdeğeriyle premedikasyon uygulanmalıdır; deksametazon hem premedikasyon hem de anti-miyelom tedavisi olarak kullanılır (bkz. Bölüm 4.2). SARCLISA infüzyonunun tamamı boyunca yaşamsal belirtiler sık sık izlenmelidir. Gerektiği zaman SARCLISA infüzyonuna ara verilmeli ve uygun tıbbi ve destekleyici önlemler alınmalıdır (bkz. Bölüm 4.2). SARCLISA infüzyonuna ara verilmesinden sonra semptomlar iyileşmezse, uygun tıbbi ürünlerle ilk iyileşmeden sonra tekrar ederse veya hastaneye yatış gerektirirse ya da yaşamı tehdit ederse, SARCLISA kalıcı olarak durdurulmalıdır ve uygun yönetim başlatılmalıdır.
Nötropeni
SARCLISA ile tedavi edilen hastalarda laboratuvar anomalileri (%84,9) ve nötropenik komplikasyonlar (%30,3) olarak Derece 3.-4 nötropeni rapor edilmiştir (bkz. Bölüm 4.8). Tedavi sırasında periyodik olarak tam kan sayımı izlenmelidir. Nötropeni hastaları enfeksiyon belirtileri açısından izlenmelidir. SARCLISA dozunun azaltılması önerilmemektedir. Nötropeni riskinin azaltılması için, SARCLISA dozunun ertelenmesi ve koloni uyarıcı faktörlerin (örn. G-CSF) kullanılması düşünülmelidir.
Enfeksiyon
SARCLISA tedavisinde, çoğunlukla pnömoni, üst solunum yolu enfeksiyonu ve bronşit olmak üzere Derece ≥3 enfeksiyonlar dahil daha yüksek enfeksiyon insidansı gerçekleşmiştir (bkz. Bölüm 4.8). SARCLISA alan hastalar enfeksiyon belirtileri açısından yakından izlenmeli ve uygun standart tedavi uygulanmalıdır. Tedavi sırasında antibiyotik, antifungal ve antiviral profilaksi düşünülebilir.
İkinci primer maligniteler
ICARIA-MM çalışmasında, SARCLISA ile tedavi edilen 6 hastada (%3,9) ve pomalidomid ve deksametazonla tedavi edilen 1 hastada (%0,7) ikinci primer maligniteler (SPM) bildirilmiştir ve bunlar arasında SARCLISA ile tedavi edilen 4 hastada ve pomalidomid ve deksametazonla tedavi edilen 1 hastada deride skuamöz hücreli karsinom bulunmaktadır (bkz. Bölüm 4.8). Hastalar deride skuamöz karsinom rezeksiyonundan sonra tedaviye devam etmiştir. SARCLISA'ya maruz kalan tüm hastalarda toplam SPM insidansı %3'tür. Doktorlar tedaviden önce ve tedavi sırasında hastaları SPM oküransı için Uluslararası Myelom Çalışma Grubu (IMWG) kılavuzlarına göre dikkatli şekilde değerlendirmeli ve endike tedaviyi başlatmalıdır.
Serolojik testlerle etkileşim (indirekt antiglobülin testi)
İsatuksimab, kırmızı kan hücresindeki (RBC) CD38'e bağlanmaktadır ve indirekt antiglobülin testinde (indirekt Coombs testi) hatalı pozitif ile sonuçlanabilmektedir. RBC transfüzyonuyla potansiyel problemlerden kaçınmak için, SARCLISA tedavisi gören hastaların kan tipi ve tarama testleri ilk infüzyondan önce yapılmalıdır. Yerel uygulamalara göre, SARCLISA tedavisine başlamadan önce fenotipleme düşünülebilir. SARCLISA tedavisi başlatılmışsa, kan bankası bilgilendirilmelidir. Hastalar teorik hemoliz riski açısından izlenmelidir. Acil bir transfüzyon gerekirse, yerel kan bankası uygulamalarına göre çapraz karşılaştırma yapılmamış ABO/Rh-uygun RBC verilebilir (bkz. Bölüm 4.5). Son SARCLISA infüzyonundan sonra indirekt Coombs testiyle etkileşimin ne kadar uzun süreceğiyle ilgili bilgi bulunmamaktadır. İsatuksimabın yarı ömrüne göre, isatuximab aracılı pozitif indirekt Coombs testinin son infüzyondan sonra yaklaşık 6 ay boyunca devam edebileceği beklenmektedir.
Tam yanıtın belirlenmesinde etkileşim
İsatuksimab, endojen M proteininin klinik izleminde kullanılan serum protein elektroforez (SPE) ve immünofiksasyon (IFE) tayinlerinde belirlenebilen bir IgG kappa monoklonal antikorudur (bkz. Bölüm 4.5). Bu etkileşim, IgG kappa miyelom proteini olan bazı hastalarda tam yanıtın belirlenmesindeki doğruluğu etkileyebilmektedir. Çok İyi Kısmi Yanıt (VGPR) kriterlerini karşılayan ve yalnızca residual immünofiksasyon pozitiflik gösteren isatuximab rejimi kolundaki 22 hasta etkileşim açısından test edilmiştir. İsatuksimab sinyalini miyelom M proteini sinyalinden ayırt etmek için kütle spektrometrisi yoluyla bu hastalardan alınan serum numuneleri test edilmiştir (bkz. Bölüm 4.5).
Yaşlı hastalar
85 yaş ve üzerindeki yaşlı popülasyonla ilgili veriler sınırlıdır (bkz. Bölüm 4.2).
İzlenebilirlik
Biyoteknolojik ürünlerin takip edilebilirliğinin sağlanması için uygulanan ürünün ticari ismi ve seri numarası mutlaka hasta dosyasına kaydedilmelidir.
4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleri
İsatuksimabın pomalidomid farmakokinetiği üzerinde etkisi bulunmamaktadır ve tam tersi de geçerlidir.
Serolojik testlerle etkileşim
CD38 proteini kırmızı kan hücrelerinin yüzeyinde eksprese edildiği için, bir anti-CD38 antikoru olan isatuximab, isatuximabla tedavi edilen hastalarda; indirekt antiglobülin testlerinde (indirekt Coombs testi), antikor tespit (tarama) testlerinde, antikor tanıma panellerinde ve anti-insan globülin (AHG) çapraz eşleştirmelerinde hatalı pozitif reaksiyonlarla kan bankalarının serolojik testlerinde etkileşim gösterebilmektedir (bkz. Bölüm 4.4). Etkileşimi azaltma yöntemleri arasında, isatuximab bağlanmasını sekteye uğratmak için reaktif kırmızı kan hücrelerinin ditiyotretol (DTT) ile tedavi edilmesi ve diğer yerel olarak valide edilmiş yöntemler bulunmaktadır. Kell Kan grubu sistemi de DTT tedavisine duyarlı olduğu için, DTT ile tedavi edilen kırmızı kan hücreleri kullanılarak aloantikor olasılığının elenmesi veya aloantikor belirlenmesinden sonra Kell-negatif birimler tedarik edilmelidir.
Serum Protein Elektroforezi ve İmmünofiksasyon Testleriyle Etkileşim
Grubu (IMWG) kriterlerine göre doğru yanıt sınıflandırmasıyla etkileşime girebilmektedir (bkz. Bölüm 4.4).
Özel popülasyonlara ilişkin ek bilgiler Pediyatrik popülasyon:
Bu popülasyona özel bir etkileşim çalışması yapılmamıştır.
Geriyatrik popülasyon:
Bu popülasyona özel bir etkileşim çalışması yapılmamıştır.
4.6. Gebelik ve laktasyon
Gebelik kategorisi: D
Çocuk doğurma potansiyeli bulunan kadınlar/Doğum kontrolü (Kontrasepsiyon) İsatuksimab ile tedavi edilen çocuk doğurma potansiyeli bulunan kadınlar tedavi sırasında ve tedavinin sonra ermesinden sonra 5 ay boyunca etkili doğum kontrolü kullanmalıdır.
Gebelik dönemi
İsatuksimabın gebe kadınlardaki kullanımıyla ilgili veri bulunmamaktadır. İsatuksimabla hayvanlarda üreme toksisitesi çalışmaları yapılmamıştır. İmmünoglobülin G1 monoklonal antikorlarının gebeliğin ilk trimestresinden sonra plasentayı geçtiği bilinmektedir. Gebe kadınlarda isatuximab kullanılması önerilmemektedir.
Laktasyon dönemi
İsatuksimabın insan sütüne geçip geçmediği bilinmemektedir. İnsan IgG'lerinin doğumdan sonraki ilk birkaç gün boyunca süte geçtiği ve kısa süre sonra düşük konsantrasyonlara indiği bilinmektedir; ancak, emzirilen bebek için doğumdan sonraki bu kısa süre sırasındaki risk göz ardı edilemez. Bu spesifik dönemde, emzirmenin çocuk için yararı ve tedavinin kadın için yararı göz önünde bulundurularak, emzirmeyi bırakma veya isatuximab tedavisini bırakma/tedaviden kaçınma arasında bir karar verilmesi gerekmektedir. Bundan sonra, klinik olarak gerekliyse isatuximab emzirme döneminde kullanılabilir.
Üreme yeteneği/Fertilite
İsatuksimabın erkek ve kadın fertilitesi üzerindeki potansiyel etkilerini belirlemek için insan veya hayvan verisi bulunmamaktadır (bkz. Bölüm 5.3).
İsatuksimabla birlikte uygulanan diğer tıbbi ürünler için, ilgili güncel kısa ürün bilgisine bakınız.
4.7. Araç ve makine kullanımı üzerindeki etkiler
SARCLISA'nın araç veya makine kullanımı üzerinde etkisi yoktur veya ihmal edilebilir düzeydedir.
4.8. İstenmeyen etkiler
Güvenlilik profilinin özeti
En sık görülen advers reaksiyonlar (≥%20); nötropeni (%46,7), infüzyon reaksiyonları (%38,2), pnömoni (%30,9), üst solunum yolu enfeksiyonu (%28,3), diyare (%25,7) ve bronşittir (%23,7).
En sık görülen ciddi advers reaksiyonlar pnömoni (%9,9) ve febril nöropenidir (%6,6).
Advers reaksiyonların tablo halinde listesi
Advers reaksiyonların sıklıkları, NCI Yaygın Toksisite Kriterleri, COSTART ve MedDRA terimleri kullanılarak tanımlanmıştır: Çok yaygın (≥1/10); yaygın (≥1/100 ila <1/10); yaygın olmayan (≥1/1.000 ila <1/100); seyrek (≥1/10.000 ila <1/1.000); çok seyrek (<1/10.000); bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor).
Her bir sıklık gruplamasında, istenmeyen etkiler, azalan ciddiyet sırasına göre sunulmaktadır. Advers reaksiyonlar, isatuximab rejim grubunda tedavi edilen hastalarda ≥%5 (tüm dereceler) veya ≥%2 (Derece ≥3) insidansa göre ve kontrol rejim (pomalidomid ve düşük dozlu deksametazon) grubuyla karşılaştırıldığında isatuximab rejim grubunda insidans oranının
≥%5 olmasına göre seçilmiştir. Atriyal fibrilasyon ve deride skuamöz hücreli karsinom terimleri klinik ilgileri sebebiyle eklenmiştir.
Tablo 3: Pomalidomid ve düşük doz deksametazonla kombinasyon halinde isatuximabla tedavi edilen multipl miyelom hastalarında bildirilen advers reaksiyonlar (ICARIA-MM çalışması)
Sistem Organ Sınıfı Tercih Edilen Terim |
Advers reaksiyon |
Sıklık | İnsidans (%) (N=152) | |
Tüm dereceler | Derece ≥3 | |||
Enfeksiyonlar ve enfestasyonlar | Pnömoni | Çok yaygın | 47 (30,9) | 40 (26,3) |
Üst solunum yolu enfeksiyonu* | Çok yaygın | 43 (28,3) | 5 (3,3) | |
Bronşit* | Çok yaygın | 36 (23,7) | 5 (3,3) | |
(Kist ve polipler de dahil olmak üzere) iyi huylu ve kötü huylu neoplazmalar | Deride skuamöz hücreli karsinom | Yaygın | 4 (2,6) | 2 (1,3) |
Kan ve lenf sistemi hastalıkları | Nötropeni | Çok yaygın | 71 (46,7) | 70 (46,1) |
Febril nötropeni | Çok yaygın | 18 (11,8) | 18 (11,8) | |
Metabolizma ve beslenme hastalıkları | İştah kaybı* | Yaygın | 15 (9,9) | 2 (1,3) |
Kardiyak hastalıklar | Atriyal fibrilasyon | Yaygın | 7 (4,6) | 3 (2,0) |
Solunum, göğüs bozuklukları ve mediastinal hastalıklar | Dispne* | Çok yaygın | 23 (15,1) | 6 (3,9) |
Gastrointestinal hastalıkları | Diyare* | Çok yaygın | 39 (25,7) | 3 (2,0) |
Bulantı* | Çok yaygın | 23 (15,1) | 0 | |
Kusma* | Çok yaygın | 18 (11,8) | 2 (1,3) | |
Araştırmalar | Kilo kaybı* | Yaygın | 10 (6,6) | 0 |
Yaralanma ve zehirlenme | İnfüzyon reaksiyonu | Çok yaygın | 58 (38,2) | 4 (2,6) |
![]()
* Derece 4 yok.
Seçili advers reaksiyonların tanımı
İnfüzyon reaksiyonları
ICARIA-MM'de, SARCLISA ile tedavi edilen 58 hastada (%38,2) infüzyon reaksiyonları bildirilmiştir. İnfüzyon reaksiyonları yaşayan hastaların tümü 1. SARCLISA infüzyonu sırasında bu reaksiyonlardan yaşamıştır ayrıca 3 hasta (%2,0) 2. infüzyonunda, 2 hasta (%1,3)
4. infüzyonunda infüzyon reaksiyonları yaşamıştır. Hastaların %3,9'unda Derece 1,
%31,6'sında Derece 2, %1,3'ünde Derece 3, %1,3'ünde Derece 4 infüzyon reaksiyonları bildirilmiştir. Tüm infüzyon reaksiyonları geri dönüşümlüdür ve infüzyonların %98'inde aynı gün içinde çözülmüştür. Derece 3 ve daha yüksek infüzyon reaksiyonlarının belirtileri ve semptomları arasında dispne, hipertansiyon ve bronkospazm bulunmaktadır.
İnfüzyon reaksiyonları nedeniyle infüzyona ara verme insidansı %28,9'dur. İnfüzyona ara verilmesine kadar geçen medyan süre 55 dakikadır.
İsatuksimab rejimi grubunda hastaların %2,6'sında infüzyon reaksiyonu nedeniyle tedavilerin bırakıldığı bildirilmiştir.
Enfeksiyonlar
ICARIA-MM'de, Derece 3 veya daha yüksek enfeksiyonların insidansı %42,8'dir. Pnömoni en yaygın olarak bildirilen şiddetli enfeksiyondur ve Derece 3; isatuximab rejimi grubundaki hastaların %21,7'sine karşı kontrol rejim (pomalidomid ve düşük doz deksametazon) grubundaki hastaların %16,1'inde, Derece 4 ise isatuximab rejimi grubundaki hastaların
%3,3'üne karşı kontrol rejim (pomalidomid ve düşük doz deksametazon) grubundaki hastaların %2,7'sinde bildirilmiştir. İsatuksimab rejimi grubundaki hastaların %2,6'sına karşı kontrol rejim grubundaki hastaların %5,4'ünde enfeksiyon nedeniyle tedavilerin bırakıldığı bildirilmiştir. İsatuksimab rejim grubundaki hastaların %3,3'ünde, kontrol rejim grubundaki hastaların %4,0'ünde fatal enfeksiyonlar bildirilmiştir.
Hematoloji laboratuvar değerleri
Tablo 4: Pomalidomid ve düşük doz deksametazonla kombinasyon halinde isatuximaba karşı pomalidomid ve düşük doz deksametazon alan hastalarda hematoloji laboratuvar anomalileri (ICARIA-MM)
Laboratuvar parametresi | SARCLISA + Pomalidomid + düşük doz deksametazon n(%) (N=152) | Pomalidomid + düşük doz Deksametazon n (%) (N=147) | ||||
| Tüm dereceler | Derece 3 | Derece 4 | Tüm dereceler | Derece 3 | Derece 4 |
Anemi | 151 (99,3) | 48 (31,6) | 0 | 145 (98,6) | 41 (27,9) | 0 |
Nötropeni | 146 (96,1) | 57 (38,8) | 46 (31,3) | |||
Lenfopeni | 140 (92,1) | 64 (42,1) | 19 (12,5) | 137 (93,2) | 52 (35,4) | 12 (8,2) |
Trombositopeni | 127 (83,6) | 22 (14,5) | 25 (16,4) | 118 (80,3) | 14 (9,5) | 22 (15,0) |
Yüzde hesaplaması için kullanılan payda, ele alınan gözlem dönemi sırasında en az 1 laboratuvar testi değerlendirmesi yapılan hastaların sayısıdır.
İmmünojenisite
ICARIA-MM (N=564) dahil olmak üzere, multipl miyelomda (MM) isatuximabın tek ajan ve kombinasyon tedavileriyle kullanıldığı 6 klinik çalışmada, tedavide ortaya çıkan ADA'ların insidansı %2,3'tür. ADA'ların isatuximabın farmakokinetiği, güvenliliği veya etkililiği üzerinde bir etkisi gözlemlenmemiştir.
Şüpheli advers reaksiyonların raporlanması
Ruhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar/risk dengesinin sürekli olarak izlenmesine olanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli advers reaksiyonu Türkiye Farmakovijilans Merkezi (TÜFAM)'ne bildirmeleri gerekmektedir. (www.titck.gov.tr; e- posta: tufam@titck.gov.tr; tel: 0 800 314 00 08; faks: 0 312 218 35 99)
4.9. Doz aşımı ve tedavisi
Belirtiler ve semptomlar
Klinik çalışmalarda doz aşımıyla ilgili bir deneyim bulunmamaktadır. Klinik çalışmalarda 20 mg/kg'a kadar intravenöz isatuximab dozları uygulanmıştır.
Yönetim
SARCLISA doz aşımı için bilinen spesifik bir antidot bulunmamaktadır. Doz aşımı durumunda, hasta advers reaksiyon belirtileri veya semptomları açısından izlenmeli ve derhal tüm uygun önlemler alınmalıdır.
5. FARMAKOLOJİK ÖZELLİKLER
5.1. Farmakodinamik özellikler
Farmakoterapötik grup: Antineoplastik ajanlar, monoklonal antikorlar ATC kodu: L01FC02
Etki mekanizması
İsatuksimab, CD38 reseptörünün spesifik bir hücre dışı epitopuna bağlanan bir IgG1 türevi monoklonal antikordur. CD38, multipl miyelom hücreleri üzerinde yüksek düzeyde eksprese edilen bir transmembran glikoproteindir.
In vitro ortamda isatuximab; antikor bağımlı hücre aracılı sitotoksisite (ADCC), antikor bağımlı hücresel fagositoz (ADCP) ve kompleman bağımlı sitotoksisite (CDC) dahil olmak üzere IgG Fc-bağımlı mekanizmalar yoluyla etki göstermektedir. İsatuksimab ayrıca Fc bağımsız bir mekanizma yoluyla apoptoz indüksiyonuyla da tümör hücrelerinin ölümünü tetikleyebilmektedir.
In vitro ortamda, isatuximab kalsiyum mobilize edici bir ajan olan siklik ADP-ribozun (cADPR) sentezi ve hidrolizini katalize eden CD38'in enzimatik aktivitesini bloke etmektedir. İsatuksimab multipl miyelom hücrelerinde hücre dışı nikotinamid adenin dinükleotidden (NAD) cADPR üretimini inhibisyona uğratmaktadır.
In vitro ortamda, isatuximab CD38 pozitif hedef tümör hücrelerinin yokluğunda NK hücreleri aktive edebilmektedir.
In vivo ortamda, isatuximab monoterapisiyle tedavi edilen hastaların periferik kanında toplam CD16 ve CD56 NK hücreleri, CD19 B hücreleri, CD4 T hücreleri ve T(CD3, CD4, CD25, CD127) mutlak sayımında bir düşüş gözlemlenmiştir.
Multipl miyelom hastalarında, SARCLISA monoterapisi T hücre reseptör repertuvarında klonal genişleme indüklemiştir; bu da adaptif bir immün yanıtı olduğuna işaret etmektedir.
In vitro ortamda isatuximab ve pomalidomid kombinasyonu, tek başına isatuximabla karşılaştırıldığında efektör hücreler (ADCC) tarafından veya doğrudan tümör hücrelerini öldürerek CD38 eksprese eden multipl miyelom hücrelerinin hücre lizisini arttırmaktadır. Farelerde insan multipl miyelom ksenogreft modeli kullanılan in vivo hayvan deneylerinde, isatuximab ve pomalidomid kombinasyonunun tek başına isatuximab veya pomalidomidle karşılaştırıldığında anti-tümör aktivitede artışla sonuçlandığını göstermiştir.
Klinik etkililik ve güvenlilik ICARIA-MM (EFC 14335)
SARCLISA'nın pomalidomid ve düşük doz deksametazonla birlikte etkililiği ve güvenliliği, relaps ve refrakter multipl miyelom hastalarında yapılan çok merkezli, çok uluslu, randomize, açık etiketli, 2 kollu bir Faz 3 çalışması olan ICARIA-MM (EFC14335) çalışmasında incelenmiştir. Hastalar daha önce lenalidomid ve bir proteazom inhibitörü dahil olmak üzere en az iki tedavi görmüş ve önceki tedavi sırasında veya tedavinin sona ermesinden sonraki 60 gün içinde hastalık progresyonu göstermiştir. Primer refrakter hastalığı olan hastalar çalışmaya dahil edilmemiştir.
Toplam 307 hasta 1:1 oranında pomalidomid ve düşük deksametazonla kombinasyon halinde SARCLISA (isatuximab rejimi, 154 hasta) veya pomalidomid ve düşük doz deksametazon (kontrol rejimi, 153 hasta) tedavisine randomize edilmiştir. Tedavi iki grupta da 28 günlük sikluslarda, hastalık ilerlemesi veya kabul edilemez toksisite meydana gelene kadar uygulanmıştır. SARCLISA 10 mg/kg, ilk siklusta haftada bir ve sonraki sikluslarda her iki haftada bir IV infüzyon olarak uygulanmıştır.
Pomalidomid 4 mg, her 28 günlük siklusun 1. gününden 21. gününe kadar günde bir kez oral yoldan alınmıştır. 28 günlük siklusun 1., 8., 15. ve 22. günlerinde düşük doz deksametazon (oral/intravenöz) 40 mg (75 yaş ve üzerindeki hastalarda 20 mg) verilmiştir.
Genel olarak, başlangıçta demografik ve hastalık karakteristikleri iki tedavi grubu arasında benzerdir ve bazı minör dengesizlikler bulunmaktadır. Medyan hasta yaşı 67'dir (aralık 36-86) ve hastaların %19,9'u 75 yaş ve üzerindedir. ECOG PS isatuximab kolundaki hastaların
%35,7'sinde ve kontrol kolundaki hastaların %45,1'inde 0; isatuximab kolundaki hastaların
%53,9'u ve kontrol kolundaki hastaların %44,4'ünde 1; isatuximab kolundaki hastaların
%10,4'ü ve kontrol kolundaki hastaların %10,5'inde 2'dir. İsatuximab kolundaki hastaların
%10,4'üne karşı kontrol kolundaki hastaların %10,5'i çalışmaya KOAH veya astım öyküsüyle girmiştir ve isatuximab ve kontrol kolunda hastaların sırasıyla %38,6'sı ve %33,3'ü böbrek yetmezliği (kreatinin klirensi <60 mL/dk./1,73 m) vardır. Çalışmanın başındaki Uluslararası Evreleme Sistemi (ISS) evresi hastaların %37,5'inde I (isatuximab kolunda %41,6, kontrol kolunda %33,3), %35,5'inde II (isatuximab kolunda %34,4, kontrol kolunda %36,6) ve
kolunda %9,1, kontrol kolunda %15,0), %8,5'inde (isatuximab kolunda %7,8, kontrol kolunda
%9,2) ve %1,6'sında (isatuximab kolunda %0,6, kontrol kolunda %2,6) sırasıyla del(17p), t(4;14) ve t(14;16) bulunmaktadır.
Geçmişteki medyan tedavi basamağı sayısı 3'tür (aralık 2-11). Tüm hastalar geçmişte bir proteazom inhibitörü almıştır, tüm hastalar geçmişte lenalidomid almıştır ve hastaların
%56,4'ü geçmişte kök hücre nakli almıştır. Hastaların büyük bölümü (%92,5) lenalidomide,
%75,9'u bir proteazom inhibitöründe, %72,6'sı hem bir immünomodülatöre ve bir proteazom inhibitörüne ve refrakterdir ve hastaların %59'u son tedavi basamağı olarak lenalidomide refrakterdir.
Medyan tedavi süresi isatuximab rejimi grubunda 41 haftaya kıyasla kontrol rejim grubunda 24 haftadır.
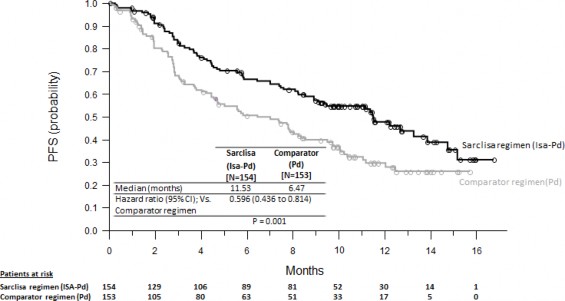
ICARIA-MM çalışmasındaki primer etkililik sonlanım noktaları progresyonsuz sağkalımdır (PFS). PFS'deki iyileşme isatuximab rejimiyle tedavi edilen hastalarda hastalık progresyonu veya ölüm riskinde %40,4 düşüş temsil etmektedir.
Etkililik sonuçları Tablo 5'te sunulmaktadır ve PFS ve Genel Sağkalım için Kaplan-Meier eğrileri Şekil 1 ve 2'de verilmiştir:
Tablo 5: Multipl miyelom tedavisinde pomalidomid ve düşük doz deksametazonla kombinasyon halinde SARCLISA'ya karşı pomalidomid ve düşük doz deksametazonun etkililiği (tedavi amaçlı analiz)
Bitiş noktası | SARCLISA + pomalidomid + düşük doz deksametazon N = 154 | Pomalidomid + düşük doz deksametazon N = 153 |
Progresyonsuz sağkalım |
|
|
Medyan (ay) | 11,53 | 6,47 |
[%95 GA] | [8,936-13,897] | [4,468-8,279] |
Tehlike oranı [%95 GA] | 0,596 [0,436-0,814] | |
p değeri (tabakalaştırılmış log- sıra testi) | 0,0010 | |
Toplam Yanıt Oranı | 93 (60,4) | 54 (35,3) |
Yanıt verenler | [0,5220-0,6817] | [0,2775-0,4342] |
(sCR+CR+VGPR+PR) n(%) |
|
|
[%95 GA] |
|
|
Olasılık oranı vs. kontrol [%95 tam GA] | 2,795 [1,715-4,562] | |
p değeri (tabakalaştırılmış Cochran-Mantel-Haenszel) | <0,0001 | |
Katı Tam Yanıt (sCR) + Tam Yanıt (CR) n(%) | 7 (4,5) | 3 (2,0) |
Çok İyi Kısmi Yanıt (VGPR) n(%) | 42 (27,3) | 10 (6,5) |
Kısmi Yanıt (PR) n(%) | 44 (28,6) | 41 (26,8) |
VGPR veya daha iyi n(%) | 49 (31,8) | 13 (8,5) |
[%95GA] | [0,2455-0,3980] | [0,0460-0,1409] |
Tehlike oranı vs. kontrol [%95 | 3k0YnUyZW56 | |
Bitiş noktası | SARCLISA + pomalidomid + düşük doz deksametazon N = 154 | Pomalidomid + düşük doz deksametazon N = 153 |
tam GA] |
| |
p değeri (tabakalaştırılmış Cochran-Mantel-Haenszel) | <0,0001 | |
Yanıt Süresi Medyan ay [%95 GA] |
13,27 [10,612-NR] |
11,07 [8,542-NR] |
NR = Ulaşılmamıştır.
Yüksek riskli sitogenetiğe sahip hastalarda (merkezi laboratuvar değerlendirmesi), medyan PFS isatuximab rejimi kolunda 7,49 (%95 GA: 2,628 ile NC), kontrol rejim grubunda 3,745'tir (%95 GA: 2,793 ile 7,885) (HR=0,655; %95 GA: 0,334 ile 1,283). 75 yaş ve
üzerindeki hastalarda da isatuximab rejimi grubunda PFS iyileşmeleri görülmüştür (HR=0,479; %95 GA: 0,242 ile 0,946) ve geçmişte lenalidomid (HR=0,593; %95 GA: 0,431
ile 0,816) veya proteazom inhibitörü (HR=0,578; %95 GA: 0,405 ile 0,824) tedavisine refrakter hastalarda ve çalışma girişinden önce son basamakta lenalidomide refrakter (HR= 0,601; %95 GA: 0,436 ile 0,828) hastalarda çalışma girişindeki ISS evresi III (HR=0,635;
%95 GA: 0,363 ile 1,110), başlangıç kreatinin klirensi <60 ml/dk./1,73 m (HR=0,502; %95
GA: 0,297 ile 0,847), geçmişte >3 basamak tedavi (HR=0,590; %95 GA: 0,356 ile 0,977) vardır.
Daha önce daratumumabla tedavi edilen hastalarda (isatuximab kolunda 1 hasta, kontrol kolunda 0 hasta) isatuximab rejiminin etkililiğiyle ilgili bir sonuca varmak için yeterli veri bulunmamaktadır.
Yanıt veren hastalarda ilk yanıta kadar geçen medyan süre isatuximab grubunda 35 güne karşı kontrol grubunda 58 gündür. Medyan izlem süresi isatuximab grubunda 11,56 ay ve kontrol grubunda 11,73 aydır, medyan toplam sağkalıma iki tedavi grubunda da ulaşılmamıştır. Genel sağkalım için tehlike oranı 0,687'dir (%95 GA: 0,461-1,023, p değeri=0,0631).
Şekil 1: PFS için Kaplan-Meier Eğrileri - ITT Popülasyonu - ICARIA-MM (IRC değerlendirmesine göre)
Şekil 2: Genel Sağkalım için Kaplan-Meier Eğrileri - ITT Popülasyonu - ICARIA-MM
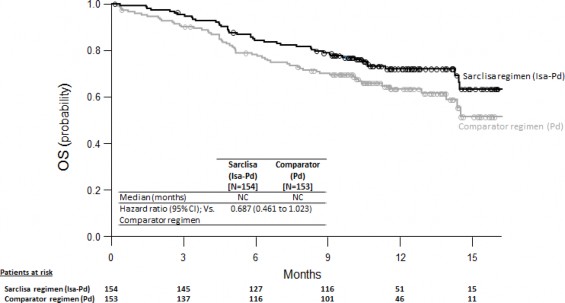
Veri sona erme tarihi = 11 Ekim 2018
ICARIA-MM (EFC14335) çalışmasında, isatuximab infüzyonu için ağırlığa dayalı bir hacim kullanılmıştır. Bölüm 4.2'de açıklanan sabit hacim infüzyonu, TCD14079 çalışmasının B
Kısmında değerlendirilmiştir ve farmakokinetik simülasyonlar hasta ağırlığına göre hacim ve
250 mL sabit hacim uygulanan enjeksiyondan sonra farmakokinetikler arasında minimal fark olduğunu teyit etmiştir (bkz. Bölüm 5.2). TCD114079 çalışmasının B kısmında, ICARIA- MM çalışmasıyla karşılaştırıldığında etkililikte hiçbir yeni güvenlilik sinyali veya etkililik farkı yoktur.
Pediyatrik popülasyon
Avrupa İlaç Ajansı (EMA) hematopoetik ve lenfoid dokulardaki malignite neoplazmların tedavisinde pediyatrik popülasyonun bir veya daha fazla alt grubunda SARCLISA ile yapılan çalışmaların sonuçlarının sunulması gerekliliğini ertelemiştir. Pediyatrik kullanımla ilgili bilgi için bkz. Bölüm 4.2.
5.2. Farmakokinetik özellikler
Genel özelliklerİsatuksimabın farmakokinetiği, tek ajan olarak veya pomalidomid ve deksametazonla kombinasyon halinde intravenöz isatuximab infüzyonuyla tedavi edilen 476 multipl miyelom hastasında, haftada bir kez, 2 haftada bir veya 8 hafta boyunca 2 haftada bir ve ardından 4 haftada bir veya 4 hafta boyunca haftada bir ve ardından 2 haftada bir olmak üzere 1 ila 20 mg/kg doz aralığında değerlendirilmiştir.
İsatuksimab maruziyeti (doz aralığında plazma konsantrasyonu-zaman eğrisi altındaki alan EAA) her 2 haftada bir programı takiben 1 mg/kg'dan 20 mg/kg'a dozla orantılı bir şekilde daha hızla artmaktayken, 4 hafta boyunca haftada bir ve ardından 2 haftada bir programı takiben 5 ve 20 mg/kg arasında doza orantılılığa sapma gözlemlenmemiştir. Bunun nedeni, 5 mg/kg'ın altındaki dozlarda doğrusal olmayan hedef aracılı klirensin toplam klirense yüksek katkısıdır ve bu katkı daha yüksek dozlarda ihmal edilebilir düzeye düşmektedir. 4 hafta boyunca haftada bir ve ardından 2 haftada bir isatuximab 10 mg/kg uygulamasından sonra, kararlı duruma ulaşana kadar geçen medyan süre 8 haftadır ve 3,1 kat birikim olmaktadır. Kararlı durumdaki ortalama (%CV) öngörülen maksimum plazma konsantrasyonu Cve EAA sırasıyla 351 mcg/mL (%36,0) ve 72.600 mcg.saat/mL'dir (%51,7). İsatuksimab infüzyonu için ağırlık bazlı bir hacim uygulama yönteminden sabit hacim infüzyonu yöntemine yapılan değişiklik, t'ta değişiklikle sonuçlanmıştır ve bu değişiklik maruziyet farmakokinetikleri üzerinde sınırlı etki göstermiştir; medyan ağırlığa (76 kg) sahip bir hasta için simülasyonu yapılan kararlı durum C(283 mcg/mL'ye 284 mcg/mL) ve 4 haftalık C(119 mcg/mL'ye 119 mcg/mL) benzerdir. Diğer hasta ağırlığı grupları için, Cve Cbenzerdir.
İsatuksimab ve pomalidomidin farmakokinetiği birlikte uygulamadan etkilenmemiştir. Emilim:
SARCLISA intravenöz olarak uygulandığı için bu bölüm geçerli değildir.
Dağılım:
İsatuksimabın tahmini toplam dağılım hacmi 8,75 L'dir.
Biyotransformasyon:
Büyük bir protein olan isatuximabın, doymayan proteolitik katabolizma prosesleriyle metabolize edilmesi beklenmektedir.
Eliminasyon:
olmayan lineer yolak. Terapötik plazma konsantrasyonları aralığında, lineer yolak baskındır ve zaman içinde %50 oranında azalarak 9,55 mL/h (0,229 L/gün) kararlı durum değerine düşmektedir. Bu değer 28 günlük bir terminal yarı ömürle ilişkilendirilmiştir.
Doğrusallık/doğrusal olmayan durum:
İsatuksimab, CD38 reseptörlerine bağlandığı için hedef aracılı ilaç eğilimiyle birlikte doğrusal olmayan farmakokinetik sergilemektedir. İsatuximab EAA'sı 2 haftada bir, 1 mg/kg ila 20 mg/kg doz aralığında (onaylanan önerilen dozun 0,1 ila 2 katı) doza orantılıdan daha fazla artmaktadır. İsatuksimab EAA'sı 4 hafta boyunca haftada bir ve ardından 2 haftada bir 5 mg/kg ila 20 mg/kg doz aralığında (onaylanan önerilen dozun 0,5 ila 2 katı) orantılı olarak artmaktadır.
Hastalardaki karakteristik özellikler
Yaş:
36 ila 85 yaşındaki 476 hastada yapılan popülasyon FK analizlerinde, <75 yaşındaki (n=406) ve ≥75 yaşındaki (n=70) hastalarda isatuximaba benzer maruziyet görülmüştür.
Cinsiyet:
207 kadın (%43,5) ve 269 erkek (%56,5) hastayla yapılan popülasyon farmakokinetik analizinde cinsiyetin isatuximabın farmakokinetiği üzerinde klinik açıdan anlamlı bir etkisi görülmemiştir.
Irk:
377 Beyaz (%79), 25 Asyalı (%5), 18 Siyah (%4) ve 33 diğer ırk (%7) hastayla yapılan popülasyon farmakokinetiği analizinde, ırkın isatuximabın farmakokinetiği üzerinde klinik açıdan anlamlı hiçbir etkisi görülmemiştir.
Ağırlık:
Kararlı durumdaki isatuximab maruziyeti (EAA) artan vücut ağırlığıyla azalmıştır.
Karaciğer yetmezliği:
Karaciğer yetmezliği olan hastalarda isatuximabla herhangi bir resmi çalışma yapılmamıştır. Popülasyon farmakokinetik analizindeki 476 hastadan 65'inde hafif karaciğer yetmezliği [toplam bilirubin normalin üst limitinin (ULN) 1 ila 1,5 katı veya aspartat amino transferaz (AST) > ULN] ve 1 hastada orta dereceli karaciğer yetmezliği (toplam bilirubin ULN'nin >1,5 ila 3 katı ve tüm AST) vardır. Hafif karaciğer yetmezliğinin isatuximabın farmakokinetiği üzerinde klinik açıdan anlamlı bir etkisi olmamıştır. Orta dereceli (toplam bilirubin ULN'nin
>1,5 ila 3 katı ve tüm AST) ve şiddetli karaciğer yetmezliğinin (toplam bilirubin ULN'nin >3 katı ve tüm AST) isatuximab farmakokinetiği üzerindeki etkisi bilinmemektedir. Ancak, isatuximab bir monoklonal antikor olduğu için, hepatik enzim aracılı metabolizma yoluyla temizlenmesi beklenmemektedir ve bu yüzden, karaciğer işlevindeki farklılıkların isatuximabın eliminasyonunu etkilemesi beklenmemektedir (bkz. Bölüm 4.2).
Böbrek yetmezliği:
Böbrek yetmezliği olan hastalarda isatuximabla herhangi bir resmi çalışma yapılmamıştır. Popülasyon farmakokinetik analizindeki 476 hasta arasında, hafif böbrek yetmezliği (60 mL/dk./1,73 m ≤ tahmini glomerüler filtrasyon oranı (eGFR) <90 mL/dk./1,73 m) olan 192 hasta, orta dereceli böbrek yetmezliği olan 163 hasta (30 mL/dk./1,73 m≤ e-GFR < 60 mL/dk./1,73 m) ve şiddetli böbrekyetmezliği o lan12hasta (e-GFR <30 mL/dk./1,73 m)
bulunmaktadır. Analizler, normal böbrek işleviyle karşılaştırıldığında hafif ila orta dereceli böbrek yetmezliğinin isatuximabın farmakokinetiği üzerinde klinik açıdan anlamlı bir etkisi olduğunu göstermemektedir.
Pediyatrik popülasyon:
İsatuksimab 18 yaşından küçük hastalarda değerlendirilmemiştir.
5.3. Klinik öncesi güvenlilik verileri
Klinik dışı veriler, tekrarlanan konvansiyonel doz toksisitesi çalışmalarına göre insanlarda özel bir tehlike ortaya koymamıştır ancak seçilen türler farmakolojik açıdan duyarlı değildir ve bu nedenle insanlardaki önemi bilinmemektedir. Genotoksisite, karsinojenik potansiyel ve üreme ve gelişim toksisite çalışmaları yapılmamıştır.
6. FARMASÖTİK ÖZELLİKLER
6.1. Yardımcı maddelerin listesi
Sükroz
Histidin hidroklorür monohidrat Histidin
Polisorbat 80 Enjeksiyonluk su
6.2. Geçimsizlikler
Bu tıbbi ürün Bölüm 6.6'da belirtilenler dışında hiçbir tıbbi ürünle karıştırılmamalıdır.
6.3. Raf ömrü
Açılmamış Flakon 36 ay
Seyreltildikten sonra
SARCLISA infüzyonunun kimyasal ve fiziksel kullanım stabilitesi, 2°C-8°C'de 48 saat ve ardından oda sıcaklığında 8 saat (infüzyon süresi dahil) boyunca kanıtlanmıştır. Mikrobiyolojik açıdan ürün hemen kullanılmalıdır. Eğer hemen kullanılmazsa, kullanımdaki saklama süreleri ve kullanımdan önceki koşullar kullanıcının sorumluluğundadır ve seyreltme kontrollü ve valide edilmiş aseptik koşullarda gerçekleştirilmediyse, normalde 2°C ila 8°C'de 24 saatten uzun olmayacaktır.
İnfüzyon torbasında saklamak için ışıktan koruma gerekmemektedir.
6.4. Saklamaya yönelik özel tedbirler
Buzdolabında (2°C-8°C'de) saklayınız. Dondurmayınız.Işıktan korumak için orijinal kutusunda saklanmalıdır.
Tıbbi ürünün seyreltilmesinden sonraki saklama koşulları için bkz. Bölüm 6.3.
6.5. Ambalajın niteliği ve içeriği
ETFE (etilen ve tetrafloroetilenin kopolimeri) kaplı bromobütil tıpayla kapalı 6 mL tip I
geçme kapağa sahip alüminyum bir contayla kapatılmıştır. Dolum hacmi (yani 5,4 mL) 5 mL çıkarılmasını temin etmek için belirlenmiştir. Ambalaj boyutu bir flakon.
Tüm ambalaj boyutları piyasada olmayabilir.
6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemler
İntravenöz uygulamanın hazırlanması
İnfüzyon çözeltisi aseptik koşullarda hazırlanmalıdır.
SARCLISA konsantresinin dozu (mg), hastanın ağırlığına göre hesaplanmalıdır (dozu uygun şekilde ayarlamak için her döngüden önce ölçüm yapılır, bkz. Bölüm 4.2). Hasta için gereken dozu elde etmek için birden fazla flakon gerekebilir.
SARCLISA konsantresi flakonları, partikül içermediğinden ve renk bozukluğu olmadığından emin olmak için seyreltmeden önce görsel olarak incelenmelidir.
Flakonları çalkalamayınız.
SARCLISA konsantresinin gereken hacmine eşit seyreltici hacmi 250 mL sodyum klorür 9 mg/mL (%0,9) enjeksiyonluk çözelti veya glikoz %5 çözelti seyreltme torbasından alınmalıdır.
SARCLISA konsantresinin uygun hacmi çekilmeli ve 250 mL sodyum klorür 9 mg/mL (%0,9) enjeksiyonluk çözelti veya glikoz %5 çözeltide seyreltilmelidir.
İnfüzyon torbası poliolefin (PO), polietilen (PE), polipropilen (PP), polivinil klorür (PVC) ve di (2-etilheksil) ftalat (DEHP) veya etil vinil asetattan (EVA) yapılmış olmalıdır.
Seyreltilen çözeltiyi torbayı tersine çevirerek nazikçe homojenize ediniz. Çalkalamayınız.
Uygulama
İnfüzyon çözeltisi, intravenöz tüplü infüzyon seti (PE, DEHP varlığında veya yoluğunca PVC, polibutadiyen (PBD) veya poliüretan (PU)) ve düz eksenli filtre (polietersülfon (PES), polisülfon veya naylon) kullanılarak intravenöz infüzyon yoluyla uygulanmalıdır.
İnfüzyon çözeltisi infüzyon oranına bağlı olan bir süre boyunca uygulanmalıdır (bkz. Bölüm 4.2).
Hazırlanan infüzyon torbasının standart yapay ışıklı bir ortamda ışıktan korunması gerekmemektedir.
SARCLISA'yı başka ajanlarla birlikte aynı intravenöz yoldan eşzamanlı olarak uygulamayınız.
 Parkinson Hastalığı
Hastalık ilk kez 1817 de İngiliz doktor James Parkinson tarafından tanımlanmış ve Dr. Parkinson hastalığı “sallayıcı felç” olarak kaleme almış.
Parkinson Hastalığı
Hastalık ilk kez 1817 de İngiliz doktor James Parkinson tarafından tanımlanmış ve Dr. Parkinson hastalığı “sallayıcı felç” olarak kaleme almış. |
 Sırt Ağrısı
Sırt ağrısı birden bire ortaya
çıkıp şiddetli (akut) olabilir veya zamanla gelişip daha uzun
süreli sorunlara (kronik) neden olabilir.
Sırt Ağrısı
Sırt ağrısı birden bire ortaya
çıkıp şiddetli (akut) olabilir veya zamanla gelişip daha uzun
süreli sorunlara (kronik) neden olabilir. |
 |
Astım Astımlı kişilerin akciğerlerindeki hava boruları (bronşlar) hassastır. Bu kişiler belirli tetikleyici faktörlere maruz kaldıklarında, hava boruları nefes almalarını güçleştirecek şekilde daralır. |
 |
Pankreas Kanseri Pankreas karnın alt kısmında yatay şekilde bulunan bir organdır. Sindirime yardımcı olan enzimleri ve kan şekerini yönetmeye yardımcı olan hormonları vücuda dağıtmakla görevlidir. |
 |
Ruh ve Akıl Sağlığımızı Geliştirmek İyi akıl ve ruh sağlığı sahip olmaktan ziyade, yaptığınız şeylerdir. Akıl ve ruhsal olarak sağlıklı olmak için kendinize değer vermeli ve kendinizi kabul etmelisiniz. |
İLAÇ GENEL BİLGİLERİ
Sanofi Sağlık Ürünleri Ltd.Şti
| Satış Fiyatı | 6128.1 TL [ 26 Apr 2024 ] |
| Önceki Satış Fiyatı | 6128.1 TL [ 22 Apr 2024 ] |
| Original / Jenerik | Original İlaç |
| Reçete Durumu | Kısıtlanmış Beyaz Reçeteli bir ilaçdır. |
| Barkodu | 8699809779266 |
| Etkin Madde | Isatuksimab |
| ATC Kodu | L01FC02 |
| Birim Miktar | 100/5 |
| Birim Cinsi | MG/ML |
| Ambalaj Miktarı | 1 |
| Antineoplastik ve İmmünomodülatör Ajanlar |
| İthal ( ref. ülke : Almanya ) ve Beşeri bir ilaçdır. |