BYRETU 250 mg tablet (120 tablet) K�sa �r�n Bilgisi
{ Abirateron }
1. BE�ER� TIBB� �R�N�N ADI
BYRETU 250 mg tablet
2. KAL�TAT�F VE KANT�TAT�F B�LE��M
Etkin madde
Her bir tablet 250 mg abirateron asetat i�erir.
Yard�mc� maddeler
Her bir tablet 150 mg laktoz monohidrat (s���r kaynakl�), 15 mg kroskarmeloz sodyum ve 20 mg sodyum lauril s�lfat i�erir.
Yard�mc� maddeler i�in B�l�m 6.1'e bak�n�z.
3. FARMAS�T�K FORMU
Tablet
Beyaz ya da beyaz�ms� oval-oblong �ekilli tabletler.
4. KL�N�K �ZELL�KLER
4.1. Terap�tik endikasyonlar
BYRETU, prednizolon ile birlikte a�a��daki durumlar�n tedavisinde endikedir:
Hormonal tedaviye duyarl� ancak kemoterapi i�in uygun olmayan* metastatik prostat kanserli hastalarda progresyona kadar;
4.2. Pozoloji ve uygulama �ekli
Pozoloji, uygulama s�kl��� ve s�resi:Bu t�bbi �r�n uygun bir sa�l�k mesle�i mensubu taraf�ndan re�ete edilmelidir.
BYRETU g�nde tek seferde 1.000 mg (d�rt adet 250 mg tablet) olarak yutulmal� ve yiyeceklerle birlikte al�nmamal�d�r (uygulama �ekli b�l�m�ne bak�n�z). BYRETU'nun yiyeceklerle birlikte al�nmas� ilaca sistemik maruziyeti artt�r�r (bkz. B�l�m 4.5 ve 5.2).
Prednizolon dozu
BYRETU, metastatik hormona duyarl� prostat kanserli hastalarda g�nde 5 mg prednizolon ile birlikte kullan�l�r.
BYRETU, metastatik kastrasyona diren�li prostat kanserli hastalarda g�nde 10 mg prednizolon ile birlikte kullan�l�r.
Cerrahi kastrasyon uygulanmam�� hastalarda tedavi s�ras�nda luteinizan hormon serbestleyici hormon (LHRH) analo�u ile t�bbi kastrasyona devam edilmelidir.
�nerilen izlem
BYRETU ile tedaviye ba�lamadan �nce serum transaminaz d�zeyleri �l��lmelidir; tedavinin ilk �� ay�nda iki haftada bir, daha sonra ayda bir bu testler tekrarlanmal�d�r. Hastalar kan bas�nc�, serum potasyumu ve s�v� retansiyonu a��s�ndan ayl�k olarak izlenmelidir. Bununla birlikte, konjestif kalp yetmezli�i a��s�ndan anlaml� bir risk ta��yan hastalar tedavinin ilk 3 ay� boyunca iki haftada bir, daha sonra ise ayda bir kere izlenmelidir (bkz. B�l�m 4.4).
Daha �nceden hipokalemisi olan ya da BYRETU tedavisi s�ras�nda hipokalemi geli�en hastalarda, hastan�n potasyum d�zeyinin 4.0 mM ve �zerinde tutulmas� de�erlendirilmelidir.
Hipertansiyon, hipokalemi, �dem ve di�er mineralokortikoid d��� toksisiteler dahil olmak �zere, Derece 3 ve �zerinde toksisite geli�en hastalarda, tedavi durdurulmal� ve uygun t�bbi tedaviye ba�lanmal�d�r. Toksisite semptomlar� Derece 1 ya da ba�lang�� d�zeyine d�n�nceye kadar BYRETU tedavisine yeniden ba�lanmamal�d�r.
BYRETU veya prednizolonun g�nl�k dozunun al�nmas�n�n unutulmas� durumunda, tedaviye ertesi g�n ola�an g�nl�k dozla devam edilmelidir.
Uygulama �ekli:
BYRETU a��zdan al�n�r.
Tabletler g�nde bir kez a� karn�na tek doz olarak al�nmal�d�r. BYRETU yemek yedikten en az iki saat sonra al�nmal� ve BYRETU'y� ald�ktan sonra en az bir saat boyunca yemek yenmemelidir. BYRETU tabletleri b�t�n olarak suyla yutulmal�d�r.
�zel pop�lasyonlara ili�kin ek bilgiler:
Hepatotoksisite
BYRETU tedavisi s�ras�nda hepatotoksisite geli�en hastalarda (alanin aminotransferaz [ALT] veya aspartat aminotransferaz [AST] d�zeylerinin normal kabul edilen �st s�n�r�n 5 kat�ndan fazla y�kselmesi) tedavi hemen durdurulmal�d�r (bkz. B�l�m 4.4). Karaci�er fonksiyon testleri tedaviye ba�lamadan �nceki ba�lang�� de�erlerine d�nd�kten sonra tedaviye azalt�lm�� dozla g�nde
500 mg (iki tablet) olarak ba�lanabilir. Tedaviye yeniden ba�lanan bu hastalarda serum transaminaz d�zeyleri, tedavinin ilk �� ay�nda iki haftada bir, daha sonra ise ayda bir �l��lmelidir. Azalt�lm�� 500 mg'l�k g�nl�k dozla da hepatotoksisite tekrarlarsa, tedavi t�m�yle kesilmelidir.
Tedavinin herhangi bir d�neminde a��r hepatotoksisite geli�mesi durumunda (ALT ya da AST d�zeylerinin normal kabul edilen �st s�n�r�n 20 kat� kadar y�kselmesi) tedaviye hemen son verilmeli ve BYRETU ile yeniden tedavi uygulanmamal�d�r.
Karaci�er yetmezli�i:
�nceden hafif �iddette karaci�er bozuklu�u olan hastalarda (Child-Pugh S�n�f A) doz ayarlamas�na gerek yoktur.
Orta �iddette karaci�er yetmezli�inin (Child Pugh s�n�f B), abirateron asetat�n oral yoldan tek doz halinde 1.000 mg al�nmas� sonras� abiraterona sistemik maruziyeti yakla��k d�rt kat artt�rd��� g�sterilmi�tir (bkz. B�l�m 5.2). Orta �iddette veya a��r karaci�er yetmezli�i (Child-Pugh s�n�f B veya C) olan hastalarda abirateron asetat�n birden fazla dozunun klinik g�venlilik ve etkilili�ini g�steren bir veri bulunmamaktad�r. Bu t�r hastalarda doz ayarlamas� �ng�r�lemez. BYRETU kullan�m� faydan�n olas� riskten a��k�a a��r bast���, orta �iddette karaci�er yetmezli�ine sahip hastalarda dikkatle de�erlendirilmelidir (bkz. B�l�m 4.2 ve 5.2). BYRETU �iddetli karaci�er yetmezli�i olan hastalarda kullan�lmamal�d�r (bkz. B�l�m 4.3, 4.4 ve 5.2).
B�brek yetmezli�i:
B�brek yetmezli�i olan hastalarda doz ayarlamas�na gerek yoktur (bkz. B�l�m 5.2). Ancak prostat kanserli a��r b�brek yetmezli�i olan hastalarda klinik deneyim bulunmamaktad�r. Bu t�r hastalarda dikkatli olunmas� tavsiye edilmektedir (bkz. B�l�m 4.4).
Pediyatrik pop�lasyon:
Pediyatrik pop�lasyonda prostat kanseri g�r�lmedi�i i�in BYRETU'nun �ocuklarda ve adolesanlarda kullan�m� bulunmamaktad�r.
Geriyatrik pop�lasyon:
Ya�l� hastalarda herhangi bir doz ayarlamas� gerekmemektedir.
4.3. Kontrendikasyonlar
Abirateron asetata veya B�l�m 6.1'de listelenen herhangi bir yard�mc� maddeye kar�� a��r� duyarl�l�k varsa,
4.4. �zel kullan�m uyar�lar� ve �nlemleri
Mineralokortikoid fazlal���na ba�l� hipertansiyon, hipokalemi, s�v� retansiyonu ve kalp yetmezli�i
BYRETU, CYP17 inhibisyonu sonucunda (bkz. B�l�m 5.1) mineralokortikoid d�zeylerindeki art��a ba�l� olarak hipertansiyon, hipokalemi ve s�v� retansiyonuna (bkz. B�l�m 4.8) yol a�abilir. BYRETU ile birlikte kortikosteroid uygulanmas�, adrenokortikotropin hormon (ACTH) salg�lanmas�n� bask�layarak bu advers etkilerin g�r�lme s�kl��� ve �iddetinde bir azalma sa�lar. Kan bas�nc�n�n y�kselmesi, hipokalemi (�rne�in kardiyak glikozidleri kullanan hastalar) veya s�v� retansiyonu (�rne�in kalp yetmezli�i olan hastalar, a��r veya stabil olmayan anjina pektoris, yak�n zamanda miyokard enfarkt�s� veya ventrik�ler aritmi ve a��r b�brek yetmezli�i) nedeniyle altta yatan t�bbi durumu risk alt�na girebilecek hastalar tedavi edilirken dikkatli olunmal�d�r.
BYRETU kardiyovask�ler hastal�k �yk�s� olan hastalarda dikkatle kullan�lmal�d�r. Hipertansiyonu kontrol alt�na al�namayan, miyokard enfarkt�s�yle ortaya ��km�� klinik a��dan anlaml� kalp hastal��� olan, son 6 ayda arteriyel trombotik olay ge�irmi� olan, a��r ya da unstabil anjinas� olan, New York Kalp Cemiyeti (NYHA) S�n�f III ve IV kalp yetmezli�i olan (�al��ma 301) ya da S�n�f II'den IV'e kalp yetmezli�i (�al��ma 3011 ve 302) olan ya da kardiyak ejeksiyon fraksiyonu %50'nin alt�nda olan hastalar Faz 3 �al��malara dahil edilmemi�tir. Medikal terapi gerektiren atriyal fibrilasyonu veya di�er kardiyak aritmisi olan hastalar �al��ma 3011 ve 302'den ��kar�lm��t�r. Sol ventrik�l ejeksiyon fraksiyonu (LVEF) %50'nin alt�nda olan hastalar ya da New York Kalp Cemiyeti (NYHA) S�n�f III ve IV kalp yetmezli�i olan (�al��ma 301'de) ya da NYHA S�n�f II'den IV'e kalp yetmezli�i (�al��ma 3011 ve 302) olan hastalarda g�venlilik de�erlendirilmemi�tir (bkz. B�l�m 4.8 ve 5.1).
Konjestif kalp yetmezli�i a��s�ndan anlaml� risk ta��yan hastalar�n tedavi edilmesinden �nce (�rne�in, kalp yetmezli�i, kontrol alt�na al�namayan hipertansiyon ya da iskemik kalp hastal��� gibi kalp olaylar�), kalp fonksiyonunun bir de�erlendirmesinin yap�lmas� d���n�lmelidir (�rne�in, ekokardiyogram). BYRETU ile tedaviden �nce, kalp yetmezli�i tedavi edilmeli ve kalp fonksiyonu m�mk�n olan en iyi d�zeye ��kar�lmal�d�r. Hipertansiyon, hipokalemi ve s�v� retansiyonu d�zeltilmeli ve kontrol alt�na al�nmal�d�r. Tedavi s�ras�nda, 3 ay s�reyle 2 haftada bir ve daha sonra ayda bir kere olmak �zere kan bas�nc�, serum potasyum, s�v� retansiyonu (kilo art���, periferik �dem) ve di�er konjestif kalp yetmezli�i bulgu ve belirtileri izlenmeli ve anormallikler d�zeltilmelidir. Abirateron asetat ile ili�kili hipokalemi g�r�len hastalarda QT uzamas� g�zlenmi�tir. Kalp fonksiyonunda klinik olarak anlaml� bir azalma olmas� durumunda, kalp fonksiyonu klinik endikasyona g�re de�erlendirilmeli, uygun tedavi ba�lat�lmal� ve BYRETU tedavisinin kesilmesi de�erlendirilmelidir (bkz. B�l�m 4.2).
Hepatotoksisite ve karaci�er yetmezli�i
Kontroll� klinik �al��malarda karaci�er enzimlerinde ilac�n kesilmesine ya da doz de�i�ikli�ine neden olan belirgin art��lar bildirilmi�tir (bkz. B�l�m 4.8). BYRETU ile tedaviye ba�lamadan
sonra ayda bir bu testler tekrarlanmal�d�r. Hepatotoksisitenin klinik belirti veya bulgular� g�r�l�r
g�r�lmez, hemen serum transaminaz d�zeyleri �l��lmelidir. Tedavinin herhangi bir yerinde ALT ya da AST d�zeyleri normal kabul edilen �st s�n�r�n (N�S) 5 kat�ndan fazla y�kselen hastalarda BYRETU tedavisine hemen ara verilmeli ve karaci�er fonksiyonlar� yak�ndan takip edilmelidir.
BYRETU ile yeniden tedaviye ancak karaci�er fonksiyon testleri ba�lang�� de�erlerine d�nd���nde ve azalt�lm�� dozlarla ba�lanabilir (bkz. B�l�m 4.2).
Tedavinin herhangi bir d�neminde a��r hepatotoksisite geli�mesi durumunda (ALT ya da AST d�zeylerinin normal kabul edilen �st s�n�r�n 20 kat� kadar y�kselmesi) BYRETU tedavisi kesilmeli ve bir daha ba�lat�lmamal�d�r.
Klinik �al��malara aktif veya semptomatik viral hepatiti olan hastalar dahil edilmemi�tir; bu nedenle BYRETU'nun bu pop�lasyonda kullan�m�n� destekleyen bir veri bulunmamaktad�r.
Orta �iddette veya a��r karaci�er yetmezli�i (Child-Pugh s�n�f B veya C) olan hastalarda abirateron asetat�n birden fazla dozunun klinik g�venlilik ve etkilili�ini g�steren bir veri bulunmamaktad�r. BYRETU kullan�m�, faydan�n olas� riskten a��k�a a��r bast���, orta �iddette karaci�er yetmezli�ine sahip hastalarda dikkatle de�erlendirilmelidir (bkz. B�l�m 4.2 ve 5.2). BYRETU �iddetli karaci�er yetmezli�i olan hastalarda kullan�lmamal�d�r (bkz. B�l�m 4.2, 4.3 ve 5.2).
Akut karaci�er yetmezli�i ve fulminant hepatit tan�mlayan az say�da pazarlama sonras� rapor mevcuttur. Bunlar�n baz�lar� �l�mle sonu�lanm��t�r (bkz. B�l�m 4.8)
Kortikosteroidin geri �ekilmesi ve stresli durumlar�n kar��lanmas�
Prednizolon tedavisinin kesilmesi durumunda dikkatli olunmas� ve adrenokortikal yetmezlik geli�memesi i�in hastalar�n izlenmesi �nerilir. Kortikosteroidler kesildikten sonra BYRETU tedavisine devam edilecekse, hastalar mineralokortikoid fazlal���na ba�l� semptomlar a��s�ndan izlenmelidir (bkz. B�l�m 4.4 Mineralokortikoid fazlal���na ba�l� hipertansiyon, hipokalemi, s�v� retansiyonu ve kalp yetmezli�i ba�l���).
Prednizolon kullanan hastalarda ola�an d��� stres ortaya ��kt���nda, bu stresli durum �ncesinde, s�ras�nda ve sonras�nda kortikosteroid dozunun artt�r�lmas� gerekebilir.
Kemik dansitesi
�leri evre metastatik prostat kanseri olan erkeklerde kemik dansitesinde azalma g�r�lebilir. BYRETU'nun bir glukokortikoid ile birlikte kullan�m� bu etkiyi artt�rabilir.
Daha �nceden ketokonazol kullan�m�
Daha �nceden prostat kanseri i�in ketokonazol kullanm�� olan hastalarda daha d���k yan�t oranlar� beklenebilir.
Hiperglisemi
Glukokortikoid kullan�m� hiperglisemiyi art�rabilece�i i�in, diyabetli hastalarda kan �ekeri s�kl�kla
reg�lasyonunu bozabilece�inden dikkatli kullan�lmal�d�r
Hipoglisemi
�nceden diyabeti olup pioglitazon veya repaglinid alan hastalara BYRETU ve prednizolon uyguland���nda hipoglisemi vakalar� bildirilmi�tir (bkz. B�l�m 4.5); bu nedenle diyabetli hastalarda kan �ekeri izlenmelidir.
Kemoterapi ile kullan�m�
Abirateron Asetat'nun sitotoksik kemoterapi ile e�zamanl� olarak kullan�lmas�n�n g�venlili�i ve etkilili�i g�sterilmemi�tir (bkz. B�l�m 5.1).
Potansiyel riskler
BYRETU ile tedavi g�renler de dahil, prostat kanseri olan erkeklerde anemi ve cinsel i�lev bozuklu�u g�r�lebilir.
�skelet kas� etkileri
Abirateron Asetat ile tedavi g�ren hastalarda miyopati ve rabdomiyoliz vakalar� bildirilmi�tir. Bu olaylar�n �o�u tedavinin ilk 6 ay�nda ortaya ��km�� ve BYRETU'nun kesilmesinden sonra d�zelmi�tir. Miyopati/rabdomiyoliz ile ili�kili oldu�u bilinen ila�larla e� zamanl� tedavi g�ren hastalarda dikkatli olunmas� �nerilir.
Di�er t�bbi �r�nler ile etkile�im
Azalm�� BYRETU maruziyeti riski nedeniyle, ba�ka bir tedavi se�ene�i olmad��� s�rece tedavi s�ras�nda g��l� CYP3A4 ind�kleyicilerinden ka��n�lmal�d�r (bkz. B�l�m 4.5).
Abirateron ve prednizolonun Ra-223 ile kombinasyonu
Ra-223 ile kombinasyon halinde abirateron ve prednizolon ile tedavi, asemptomatik veya hafif semptomatik prostat kanseri hastalarda y�r�t�len klinik �al��malarda g�zlendi�i �zere artm�� k�r�k riski ve artm�� mortalite e�ilimi nedeniyle kontrendikedir (bkz. B�l�m 4.3).
BYRETU'nun prednizolon ile kombine kullan�ld��� son dozunu takiben en az 5 g�n s�reyle ard���k tedavide Ra-223 ba�lanmas� �nerilmemektedir.
Yard�mc� maddeler
Bu t�bbi �r�n laktoz ihtiva eder. Nadir kal�t�msal galaktoz intolerans�, Lapp laktaz yetmezli�i ya da glukoz-galaktoz malabsorbsiyon problemi olan hastalar�n bu ilac� kullanmamalar� gerekir.
Bu t�bbi �r�n�n d�rt tabletlik her bir dozu 1 mmol'den (veya 30,3 mg'dan) fazla sodyum ihtiva eder. Bu durum kontroll� sodyum diyetinde olan hastalarda g�z �n�nde bulundurulmal�d�r.
4.5. Di�er t�bbi �r�nler ile etkile�imler ve di�er etkile�im �ekilleri
Yiyeceklerin abirateron asetat �zerindeki etkisi
BYRETU'nun yiyeceklerle birlikte al�nmas� abirateron asetat�n emilimini anlaml� derecede artt�r�r. BYRETU'nun yiyeceklerle birlikte al�nmas� halindeki etkililik ve g�venlili�i g�sterilmemi�tir, bu
4.5. Di�er t�bbi �r�nler ile etkile�imler ve di�er etkile�im �ekilleri
Di�er ila�lar�n abirateron maruziyetlerini etkileme potansiyeli
G��l� bir CYP3A4 ind�kleyicisi olan rifampisin ile �nce 6 g�n boyunca g�nde bir kere 600 mg dozunda tedavi edilen ve takiben tek bir 1.000 mg'l�k abirateron asetat dozu verilen sa�l�kl� g�n�ll�lerde y�r�t�len bir klinik farmakokinetik etkile�im �al��mas�nda, ortalama abirateron plazma EAA de�eri %55 azalm��t�r.
Ba�ka bir tedavi se�ene�i olmad��� s�rece tedavi s�ras�nda g��l� CYP3A4 ind�kleyicilerinden ka��n�lmal�d�r (�rne�in; fenitoin, karbamazepin, rifampisin, rifabutin, rifapentin, fenobarbital, St. John's wort (sar� kantaron - Hypericum perforatum).
Sa�l�kl� g�n�ll�lerde y�r�t�len ayr� bir klinik farmakokinetik etkile�im �al��mas�nda, g��l� bir CYP3A4 inhibit�r� olan ketokonazol�n e�zamanl� uygulamas� abirateronun farmakokineti�i �zerinde klinik olarak anlaml� bir etki yaratmam��t�r.
Abirateronun di�er ila�lar�n maruziyetini etkileme potansiyeli
Abirateron, hepatik ila� metabolize eden CYP2D6 ve CYP2C8 enzimlerinin bir inhibit�r�d�r. Abirateron asetat�n (art� prednizolon) tek dozda al�nan CYP2D6 substrat� dekstrometorfan �zerindeki etkisini belirlemek i�in yap�lan bir �al��mada, dekstrometorfan�n sistemik maruziyetinin (EAA) yakla��k 2,9 kat artt��� bildirilmi�tir. Dekstrometorfan�n aktif metaboliti dekstrorfan�n EAA'� ise yakla��k %33 artm��t�r.
BYRETU, �zellikle dar terap�tik indekse sahip ila�lar olmak �zere, CYP2D6 taraf�ndan aktive veya metabolize edilen ila�larla birlikte al�nd���nda dikkatli olunmal�d�r. CYP2D6 taraf�ndan metabolize edilen dar terap�tik indekse sahip ila�lar�n dozunun azalt�lmas� de�erlendirilmelidir. CYP2D6 taraf�ndan metabolize edilen ila�lara �rnek olarak metoprolol, propranolol, desipramin, venlafaksin, haloperidol, risperidon, propafenon, flekanid, kodein, oksikodon ve tramadol g�sterilebilir (kodein, oksikodon ve tramadolun aktif analjezik metabolitlerinin olu�abilmesi i�in CYP2D6 gereklidir).
Sa�l�kl� g�n�ll�ler �zerinde yap�lan bir CYP2C8 ila� etkile�im �al��mas�nda, pioglitazon 1.000 mg'l�k tek doz abirateron asetat ile birlikte verildi�inde, pioglitazonun EAA de�eri %46 artm�� ve pioglitazonun aktif metabolitleri olan M-III ve M-IV i�in EAA de�erleri %10 azalm��t�r. CYP2C8 substrat� olan dar terap�tik aral�kl� ila�lar ile birlikte BYRETU kullan�lmas� durumunda hastalar toksisite semptomlar� a��s�ndan izlenmelidir. CYP2C8 taraf�ndan metabolize edilen t�bbi �r�n �rnekleri aras�nda pioglitazon ve repaglinid bulunmaktad�r. (bkz. B�l�m 4.4)
�n vitro ortamda, abirateronun maj�r metabolitleri olan abirateron s�lfat ve N-oksit abirateron sulfat�n karaci�er al�m ta��y�c�s� OATP1B1'i inhibe etti�i ve sonu� olarak OATP1B1 taraf�ndan elimine edilen ila�lar�n konsantrasyonlar�n� art�rabilece�i g�sterilmi�tir. Ta��y�c� ba�l� temelli etkile�imi do�rulayacak klinik veri yoktur.
QT aral���n� uzatt��� bilinen ila�lar ile kullan�m�
ind�kleyebilecek ya da s�n�f III (�rn. amiadoron, sotalol, dofetilid, ibutilid) gibi antiaritmatik �r�nler,
metadon, moksifloksasin, antipsikotikler gibi �r�nler ile verilirken dikkatli olunmas� �nerilir.
Spironolakton ile kullan�m
Spironolakton androjen resept�r�ne ba�lan�r ve prostat spesifik antijen (PSA) seviyelerini art�rabilir. BYRETU ile kullan�m� �nerilmez (bkz. B�l�m 5.1).
�zel pop�lasyonlara ili�kin ek bilgiler
Herhangi bir etkile�im �al��mas� yap�lmam��t�r.
Pediyatrik pop�lasyon:
Herhangi bir etkile�im �al��mas� yap�lmam��t�r.
4.6. Gebelik ve laktasyon
Gebelik kategorisi: X
BYRETU kad�nlarda kullan�m� olan bir ila� de�ildir. BYRETU gebe olan ya da gebe olma olas�l��� bulunan kad�nlarda kontrendikedir (bkz. B�l�m 4.3 ve 5.3).
�ocuk do�urma potansiyeli bulunan kad�nlar
Gebelikte BYRETU kullan�m�na ili�kin klinik bir veri mevcut de�ildir ve BYRETU �ocuk do�urma potansiyeli bulunan kad�nlarda kullan�m i�in bir ila� de�ildir.
Do�um kontrol� (kontrasepsiyon)
Abirateron Asetat'�n ya da metabolitlerinin semen ile salg�lan�p salg�lanmad��� bilinmemektedir. Hastan�n gebe bir kad�nla cinsel ili�kiye girmesi durumunda kondom kullanmas� gerekir. Hastan�n �ocuk do�urma potansiyeli bulunan bir kad�nla cinsel ili�kiye girmesi durumunda etkili bir do�um kontrol y�ntemine ek olarak kondom kullanmas� gerekir. Hayvanlarda yap�lan �al��malar �reme toksisitesi oldu�unu g�stermi�tir (bkz. B�l�m 5.3).
Gebelik d�nemi
BYRETU kad�nlarda kullan�m� olan bir ila� de�ildir ve gebelik d�neminde ya da gebelik potansiyeline sahip kad�nlarda kontrendikedir (bkz. B�l�m 4.3 ve 5.3).
Laktasyon d�nemi
BYRETU kad�nlarda kullan�m� olan bir ila� de�ildir.
�reme yetene�i / Fertilite
Abirateron erkek ve di�i s��anlarda fertiliteyi etkilemi� olmakla birlikte bu etkiler tamamen geri d�n���ml� olmu�tur (bkz. B�l�m 5.3).
4.7. Ara� ve makine kullan�m� �zerindeki etkiler
BYRETU'nun ara� ve makine kullan�m� yetene�i �zerinde etkisi yoktur ya da ihmal edilebilir d�zeydedir.
4.8. �stenmeyen etkiler
G�venlilik profilinin �zeti
Birle�tirilmi� Faz 3 �al��malar�n�n istenmeyen yan etkiler �zerindeki bir analizinde, BYRETU ile hastalar�n %10'unda veya fazlas�nda g�zlemlenen istenmeyen etkiler; periferik �dem, hipokalemi, hipertansiyon, idrar yolu enfeksiyonu ve alanin aminotransferaz d�zeylerinde y�kselme ve/veya aspartat aminotransferaz d�zeylerinde y�kselmedir.
Di�er �nemli advers etkiler aras�nda kalp hastal�klar�, hepatotoksisite, k�r�klar ve alerjik alveolit bulunur.
Abirateron Asetat, etki mekanizmas�n�n farmakodinamik sonucu olarak hipertansiyon, hipokalemi ve s�v� retansiyonuna neden olabilir. Faz 3 �al��malar�nda, abirateron asetat ile tedavi edilen hastalarda, plasebo ile tedavi edilen hastalara oranla beklenen mineralokortikoid advers etkiler daha yayg�n olarak g�r�lm��t�r. �al��mada hipokalemi abirateron asetat alanlarda %18 iken plasebo alanlarda %8, hipertansiyon abirateron asetat alanlarda %22 iken plasebo alanlarda %16 ve s�v� retansiyonu (periferik �dem) abirateron asetat alanlarda %23 iken plasebo alanlarda %17 olarak bildirilmi�tir. Abirateron asetat ile tedavi edilen hastalarda, Grade 3 ve 4 hipokalemi (CTCAE, versiyon 4.0 s�n�flamas�na g�re) %6 iken plasebo alanlarda %1; abirateron asetat ile tedavi edilen hastalarda Grade 3 ve 4 hipertansiyon (CTCAE, versiyon 4.0 s�n�flamas�na g�re) %7 iken plasebo alanlarda %5; abirateron asetat ile tedavi edilen hastalarda Grade 3 ve 4 s�v� retansiyonu (periferal �dem) %1 iken plasebo alanlarda %1 olarak g�zlemlenmi�tir. Mineralokortikoid reaksiyonlar t�bbi tedaviyle genellikle ba�ar�yla y�netilebilmi�tir. Kortikosteroidlerin birlikte kullan�lmas� bu advers ila� reaksiyonlar�n�n s�kl�k ve �iddetini azalt�r (bkz. B�l�m 4.4).
Advers reaksiyonlar�n �zeti
Luteinizan hormon salg�lay�c� hormon (LHRH) analo�unun kullan�lmakta oldu�u ya da daha �nceden or�iektomi tedavisi uygulanm�� ileri evre metastatik prostat kanserli hastalardaki �al��malarda, BYRETU, d���k doz prednizolon (endikasyona ba�l� olarak g�nl�k 5 veya 10 mg) ile kombine olarak g�nde 1.000 mg dozunda kullan�lm��t�r.
Klinik �al��malarda ve pazarlama sonras� deneyimlerde g�zlenen advers etkiler a�a��daki s�kl�k derecelerine g�re listelenmi�tir. �ok yayg�n (≥1/10); yayg�n (≥1/100 ila <1/10); yayg�n olmayan (≥1/1.000 ila <1/100); seyrek (≥1/10.000 ila <1/1.000); �ok seyrek (<1/10.000), bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor).
Her bir s�kl�k grubunda, istenmeyen etkiler azalan ciddiyet s�ras�na g�re sunulmu�tur.
Enfeksiyonlar ve enfestasyonlar
�ok yayg�n: �drar yolu enfeksiyonu Yayg�n: Sepsis
Ba����kl�k sistemi hastal�klar�
Bilinmiyor: Anafilaktik reaksiyonlar
Yayg�n olmayan: Adrenal yetmezlik
Metabolizma ve beslenme hastal�klar�
�ok yayg�n: Hipokalemi Yayg�n: Hipertrigliseridemi
Kardiyak hastal�klar
Yayg�n: Kalp yetmezli�i*, anjina pektoris, atrial fibrilasyon, ta�ikardi Yayg�n olmayan: Di�er aritmiler
Bilinmiyor: Miyokard enfarkt�s, QT uzamas� (bkz. B�l�m 4.4 ve 4.5)
Vask�ler hastal�klar
�ok yayg�n: Hipertansiyon
Solunum, g���s bozukluklar� ve mediastinal hastal�klar
Seyrek: Alerjik alveolit
Gastrointestinal hastal�klar
�ok yayg�n: Diyare Yayg�n: Dispepsi
Hepato-bilier hastal�klar
�ok yayg�n: Alanin aminotransferaz d�zeylerinde y�kselme ve/veya aspartat aminotransferaz
d�zeylerinde y�kselme
Seyrek: Fulminant hepatit, akut karaci�er yetmezli�i
Deri ve deri alt� doku hastal�klar�
Yayg�n: D�k�nt�
Kas-iskelet bozukluklar�, ba� doku ve kemik hastal�klar�
Yayg�n olmayan: Miyopati, rabdomiyoliz B�brek ve idrar yolu hastal�klar� Yayg�n: Hemat�ri
Genel bozukluklar ve uygulama b�lgesine ili�kin hastal�klar
�ok yayg�n: Periferik �dem
Yaralanma ve zehirlenme
Yayg�n: K�r�klar**
* Kalp yetmezli�i ayn� zamanda konjestif kalp yetmezli�i, sol ventrik�ler disfonksiyon ve ejeksiyon fraksiyonunda azalmay� da i�ermektedir.
**K�r�klar, osteoporozu ve patolojik k�r�k d���ndaki t�m k�r�klar� i�erir.
Pazarlama sonras� deneyimden spontan bildirimler
Alanin aminotransferaz d�zeyindey�kselmeve/veyaaspartat aminotransferaz d�zeyinde
fonksiyonunu i�erir.
Abirateron asetat ile tedavi edilen hastalarda a�a��daki Grade 3 advers ila� reaksiyonlar� (CTCAE, versiyon 4.0 s�n�flamas�na g�re) g�r�lm��t�r: %5 hipokalemi; %2 idrar yolu enfeksiyonu; %4 alanin aminotransferaz d�zeyinde y�kselme ve/veya aspartat aminotransferaz d�zeyinde y�kselme,
%6 hipertansiyon, %2 k�r�k; %1 periferik �dem, kalp yetmezli�i ve atrial fibrilasyon. CTCAE (versiyon 4.0) Grade 3 hipertrigliseridemi ve anjina pektoris hastalar�n %1'inden az�nda meydana gelmi�tir. Hastalar�n %1'inden az�nda Grade 4 idrar yolu enfeksiyonu, alanin minotransferaz d�zeyinde y�kselme ve/veya aspartat aminotransferaz d�zeyinde y�kselme, hipokalemi, kalp yetmezli�i, atriyal fibrilasyon ve k�r�klar g�r�lm��t�r.
Hormona duyarl� pop�lasyonda (�al��ma 3011) hipertansiyon ve hipokalemi daha y�ksek bir insidansta g�zlemlenmi�tir. Hormona duyarl� pop�lasyonda (�al��ma 3011) hastalar�n %36,7'sinde hipertansiyon rapor edilmi�ken, �al��ma 301 ve 302'de s�ras�yla %11,8 ve %20,2'idi. Hormona duyarl� pop�lasyonda hastalar�n %20,4'�nde hipokalemi g�zlemlenmi�ken (�al��ma 3011), �al��ma 301 ve 302'de s�ras�yla %19,2 ve %14,9'idi.
ECOG2 performans durum skoru temel olan alt grup hastalarda ve ayr�ca ya�l� hastalarda (≥75 ya�), yan etkilerin g�r�lme insidans� ve d�zeyi daha y�ksekti.
Se�ilmi� advers reaksiyonlar�n tan�mlanmas� Kardiyovask�ler etkiler
Abirateron asetat ile y�r�t�len �� Faz 3 �al��mas�nda da, hipertansiyonu kontrol alt�na al�namayan, miyokard enfarkt�s�yle ortaya ��km�� klinik a��dan anlaml� kalp hastal��� olan, son 6 ayda arteriyel trombotik olay ge�irmi� olan, a��r ya da unstabil anjinas� olan, New York Kalp Cemiyeti (NYHA) S�n�f III ve IV kalp yetmezli�i (�al��ma 301) ya da S�n�f II ila IV kalp yetmezli�i (�al��ma 3011 ve 302) olan ya da kardiyak ejeksiyon fraksiyon �l��m� %50'den d���k olan hastalar �al��maya dahil edilmemi�tir. �al��maya al�nan diyabet, miyokard enfarkt�s�, serebrovask�ler olay ve ani kardiyak �l�m riski olan t�m hastalara (hem aktif ila�, hem de plasebo alanlar) ayn� zamanda �ncelikle LHRH analoglar� kullan�larak androjen azaltma tedavisi uygulanm��t�r. Faz 3 �al��mas�nda kardiyovask�ler advers reaksiyonlar�n s�kl��� ABYTUBYRETU alanlarda ve plasebo alanlarda a�a��daki gibi bulunmu�tur: Atriyal fibrilasyon %2,6 ve %2,0; ta�ikardi, %1,9 ve %1,0; anjina
pektoris %1,7 ve %0,8; kalp yetmezli�i %0,7 ve %0,2 ve aritmi, %0,7 ve %0,5.
Hepatotoksisite
Abirateron asetat ile tedavi edilen hastalarda ALT, AST ve total bilirubin d�zeylerinde y�kselmeyle seyreden hepatotoksisite bildirilmi�tir. Faz 3 klinik �al��malar�nda abirateron asetat alan hastalar�n yakla��k %6's�nda tipik olarak tedaviye ba�lad�ktan sonraki ilk 3 ayda Grade 3 ve 4 karaci�er toksisitesi (�rne�in, ALT ve AST d�zeylerinde normal kabul edilen �st s�n�r�n 5 kat�ndan fazla y�kselme veya bilirubin d�zeylerinde normal kabul edilen �st s�n�r�n 1,5 kat�ndan fazla y�kselme) bildirilmi�tir. �al��ma 3011'de, abirateron asetat ile tedavi edilen hastalar�n %8,4'�nde Grade 3 ve
4 hepatotoksisite g�zlemlenmi�tir. Abirateron asetat alan hastalardan onunda hepatotoksisite sebebiyle tedavi sonland�r�lm��t�r; bunlardan ikisi Grade 2 hepatotoksisite, alt� tanesi Grade 3 hepatotoksisite ve 2 tanesi Grade 4 hepatotoksisitedir. �al��ma 3011'de hepatotoksisite sebebiyle
�len hasta olmam��t�r. Faz 3 klinik �al��malar�nda, ba�lang�� ALT veya AST d�zeyleri y�ksek olan
daha y�ksek olmu�tur.
ALT veya AST d�zeylerinde normal kabul edilen �st s�n�r�n 5 kat�ndan fazla y�kselme oldu�unda ya da bilirubin d�zeylerinde normal kabul edilen �st s�n�r�n 3 kat�ndan fazla y�kselme oldu�unda abirateron asetat tedavisine ara verilmi� ya da kesilmi�tir. �ki olguda karaci�er fonksiyon testlerinde belirgin y�kselmeler g�r�lm��t�r (bkz. B�l�m 4.4). Ba�lang�� de�erleri normal olan bu iki hastada ALT veya AST d�zeyleri normal �st s�n�r de�erlerin 15 ila 40 kat� ve bilirubin d�zeyleri ise normal �st s�n�r de�erlerin 2 ila 6 kat� y�kselmi�tir. Abirateron asetat tedavisinin kesilmesinden sonra, her iki hastada da karaci�er fonksiyon testleri normale d�nm�� ve hastalardan birinde bu defa y�kselme olmaks�z�n abirateron asetat ile yeniden tedavi uygulanabilmi�tir. �al��ma 302'deki abirateron asetat ile tedavi edilen 35 (%6,5) hastada, Grade 3 veya 4 ALT ya da AST y�kselmeleri g�zlemlenmi�tir. Aminotransferaz y�kselmeleri 3 hasta (son abirateron asetat dozundan yakla��k 3 hafta sonra yeni �oklu karaci�er metastaz� olan 2 ve AST y�kselmesi olan 1 hasta) hari� t�m hastalarda normale d�nm��t�r. Faz 3 klinik �al��malar�nda, ALT ve AST art��lar� veya karaci�er fonksiyonlar�nda anormallik nedeniyle tedaviyi kesme oranlar� abirateron asetat alan hastalar i�in %1,1, plasebo alan hastalar i�in ise %0,6 olarak bildirilmi�tir; hepatotoksisite olaylar�na ba�l� hi�bir �l�m vakas� bildirilmemi�tir.
Klinik �al��malarda ba�lang��ta hepatiti ya da karaci�er fonksiyon testlerinde anlaml� anormallikleri olan hastalar hari� tutulmak suretiyle hepatotoksisite riski azalt�lm��t�r. �al��ma 3011'de, ba�lang�� ALT ve AST de�erleri N�S'�n 2,5 kat�ndan fazla olan hastalar, bilirubin de�erleri normal kabul edilen �st s�n�r�n 1,5 kat�ndan fazla olan hastalar ve aktif veya semptomatik viral hepatit veya kronik karaci�er hastalar�; karaci�er fonksiyon bozuklu�una ba�l� assit veya kanama bozuklu�u olan hastalar �al��maya al�nmam��t�r. �al��ma 301'de, ba�lang�� ALT ve AST d�zeyleri karaci�er metastaz�n�n olmad��� durumlarda normal kabul edilen �st s�n�r�n ≥2,5 kat�, metastaz olanlarda ise normal kabul edilen �st s�n�r�n >5 kat� olan hastalar �al��maya dahil edilmemi�tir. �al��ma 302'de, karaci�er metastaz� olanlar ve ba�lang�� ALT ve AST d�zeyleri ≥ 2,5 x N�S olan hastalar �al��maya al�nmam��t�r.
Klinik �al��maya al�nan hastalarda karaci�er fonksiyon testlerinde anormalle�me oldu�unda ise, bu hastalar�n tedavileri kesilmi� ve ancak karaci�er fonksiyon testleri tedavinin ba�lang�c�ndaki d�zeylerine d�nd�kten sonra yeniden tedavi almalar�na izin verilmi�tir (bkz. B�l�m 4.2). ALT ve AST d�zeyleri normal kabul edilen �st s�n�r�n 20 kat�ndan fazla y�kselen hastalarda yeniden tedavi uygulanmam��t�r. Bu t�r hastalarda tedaviye yeniden ba�laman�n g�venlili�i bilinmemektedir. Abirateron asetat tedavisi s�ras�nda geli�en hepatotoksisitenin mekanizmas� bilinmemektedir.
��pheli advers reaksiyonlar�n raporlanmas�
Ruhsatland�rma sonras� ��pheli ila� advers reaksiyonlar�n�n raporlanmas� b�y�k �nem ta��maktad�r. Raporlama yap�lmas�, ilac�n yarar/risk dengesinin s�rekli olarak izlenmesine olanak sa�lar. Sa�l�k mesle�i mensuplar�n�n herhangi bir ��pheli advers reaksiyonu T�rkiye Farmakovijilans Merkezi (T�FAM)'ne bildirmeleri gerekmektedir (www.titck.gov.tr; e-posta: tufam@titck.gov.tr; tel: 0 800 314 00 08; faks: 0 312 218 35 99).
4.9. Doz a��m� ve tedavisi
Abirateron Asetat ile doz a��m�na dair insan deneyimi s�n�rl�d�r.
Spesifik antidotu yoktur. Doz a��m� durumunda BYRETU uygulamas� durdurularak aritmilerin, hipokalemi ve s�v� retansiyonunun bulgu ve belirtilerinin izlenmesi de dahil olmak �zere genel destekleyici �nlemler al�nmal�d�r. Karaci�er fonksiyonlar� da de�erlendirilmelidir.
5. FARMAKOLOJ�K �ZELL�KLER
5.1. Farmakodinamik �zellikler
Farmakoterap�tik grup: Endokrin tedavisi, di�er hormon antanaloglar� ve ili�kili ajanlar ATC kodu: L02BX03
Etki mekanizmas�
Abirateron asetat in vivo olarak bir androjen biyosentez inhibit�r� olan abiraterona d�n���r. Spesifik olarak abirateron 17α-hidroksilaz/C17,20-liyaz (CYP17) enzimini se�ici olarak inhibe eder. Bu enzim testik�ler, adrenal ve prostatik t�m�r dokular�nda eksprese olur ve androjenin biyosentezi i�in gereklidir. CYP17 enzimi, s�ras�yla 17α-hidroksilasyon ve C17,20 ba��n�n k�r�lmas�yla pregnenolon ve progesteronun testesteron prek�rs�rleri olan DHEA ve androstenediona d�n���m�n� katalize eder. CYP17 inhibisyonu ayn� zamanda adrenaller taraf�ndan mineralokortikoid �retiminde art��a da yol a�ar (bkz. B�l�m 4.4).
Androjene duyarl� prostat karsinomu, androjen d�zeylerini azaltan tedaviye yan�t verir. LHRH analoglar� ya da or�iektomi gibi androjen azalt�c� tedaviler, testislerdeki androjen �retimini azaltmalar�na ra�men, adrenaller ya da t�m�r dokusundaki androjen �retimini etkilemezler. LHRH analoglar� (ya da or�iektomi) ile birlikte BYRETU tedavisi uyguland���nda serum testesteron d�zeyleri (ticari testlerle �l��ld���nde) saptanabilir d�zeylerin alt�na d��er.
Farmakodinamik etkiler
Abirateron Asetat, serum testesteron ve di�er androjen seviyelerini, tek ba��na LHRH analoglar� ya da or�iektomi ile elde edilen seviyelerin alt�na d���r�r. Bu, androjen biyosentezi i�in gerekli olan CYP17 enziminin selektif olarak inhibe edilmesinin bir sonucudur. Prostat spesifik antijen (PSA) prostat kanserli hastalarda bir biyog�sterge olarak kullan�l�r. Daha �nce taksanlarla yap�lan Abirateron asetat ile tedavi edilen hastalar�n %38'inde ba�lang�� PSA de�erlerine g�re en az %50 azalma sa�lanabilmi�ken, bu azalma oran� plasebo ile tedavi edilenlerin ancak %10'unda sa�lanabilmi�tir.
Klinik etkililik ve g�venlilik
Abirateron asetat�n etkilili�i metastatik hormona duyarl� prostat kanseri (mHDPK) ve metastatik kastrasyona diren�li prostat kanseri (mKDPK) olan hastalarda ger�ekle�tirilen plasebo kontroll� �ok merkezli randomize �� Faz 3 �al��mayla (�al��ma 3011, 302 ve 301) g�sterilmi�tir. �al��ma 3011'e, y�ksek riskli prognostik fakt�rlere sahip yeni tan� alm�� (randomizasyondan �nceki 3 ay i�erisinde) mHDPK'li hastalar dahil edilmi�tir. Y�ksek riskli prognoz a�a��daki 3 risk fakt�r�nden en az 2'sine
sahip olmak olarak tan�mlanm��t�r: (1) Gleason skorunun ≥8 olmas�; (2) kemik taramas�nda 3 veya
tedavi kolunda, standart tedavi olan ADT'ye (LHRH analo�u veya or�iektomi) ilave olarak,
Abirateron asetat g�nde 1.000 mg dozunda, g�nde tek doz 5 mg d���k doz prednizon ile kombine olarak uyguland�. Kontrol kolundaki hastalara Abirateron asetat ve prednizon yerine ADT ve plasebo verildi. �al��ma 301'e daha �nceden dosetaksel kullanm�� hastalar, �al��ma 302'ye ise daha �nce dosetaksel kullanmam�� hastalar dahil edilmi�tir. Hastalar bir LHRH analo�u kullan�yorlard� ya da daha �nce or�iektomi olmu�lard�. Aktif tedavi uygulanan kolda, Abirateron asetat, g�nde iki defa 5 mg d���k doz prednizon ya da prednizolonla kombine olarak g�nde 1.000 mg dozunda kullan�lm��t�r. Kontrol grubundaysa plaseboya ek olarak g�nde iki defa 5 mg d���k doz prednizon ya da prednizolon.uygulanm��t�r.
Serum PSA konsantrasyonlar�ndaki de�i�iklikler ba��ms�z olarak her zaman klinik fayday� g�stermeyebilir. Bu nedenle, t�m �al��malarda hastalar�n a�a��da verilen tedavi kesilme kriterlerini kar��lamalar�na kadar tedaviye devam etmeleri �nerilir.
Spironolakton androjen resept�r�ne ba�land��� ve PSA d�zeylerini art�rabilece�i i�in, t�m �al��malarda spironolakton kullan�m�na izin verilmemi�tir.
�al��ma 3011 (yeni tan� alm�� y�ksek riskli mHDPK hastalar�)
�al��ma 3011'e (n=1.199) dahil edilen hastalar�n medyan ya�� 67 idi. Abirateron asetat ile tedavi edilen hastalar�n 832'si (%69,4) beyaz �rka mensup, 246's� (%20,5) Asyal�, 25'i (%2,1) Siyahi veya Afro Amerikan, 80'i (%6,7) di�er, 13'� (%1,1) bilinmeyen/raporlanmam�� ve 3'� (%0,3) Amerikan yerlisi veya Alaska yerlisi idi. Hastalar�n %97'si i�in ECOG performans durumu 0 veya 1 idi. Bilinen beyin metastaz�, kontrol alt�na al�namayan hipertansiyonu, �nemli kalp hastal��� olan veya NYHA S�n�f II-IV kalp yetmezli�i olan hastalar �al��maya al�nmam��t�r. Metastatik hastal�ktan kaynaklanan semptomlar�n tedavisi i�in 3 aya kadar ADT veya 1 k�r palyatif radyasyon veya operasyon tedavisi alan hastalar haricinde daha �nce farmakoterapi, radyasyon terapisi veya metastatik prostat kanseri operasyonu ge�irerek tedavi edilen hastalar �al��maya al�nmam��t�r. Ortak birincil etkililik sonlan�m noktalar� genel sa�kal�m (OS) ve radyografik progresyonsuz sa�kal�m (rPFS) idi. K�sa A�r� Envanteri K�sa Form (BPI-SF) ile �l��len medyan ba�lang�� a�r� skoru hem tedavi kolunda, hem de plasebo gruplar�nda 2,0 idi. Ortak birincil sonlan�m noktalar� �l��mlerine ilave olarak; iskeletle ili�kili olaya (SRE) kadar ge�en s�re, prostat tedavisi i�in sonraki tedaviye kadar ge�en s�re, kemoterapi ba�lang�c�na kadar ge�en s�re, a�r� progresyonuna kadar ge�en s�re ve PSA progresyonuna kadar ge�en s�re kullan�larak tedavi faydas� da de�erlendirildi. Tedavi hastal�k progresyonuna, onam�n geri �ekilmesine, kabul edilemez toksisite veya �l�me kadar devam etti.
Radyografik progresyonsuz sa�kal�m, randomizasyondan radyografik progresyon g�r�lmesine veya herhangi bir nedene ba�l� �l�me kadar ge�en s�re olarak tan�mland�. Radyografik progresyon, kemik taramas�yla progresyonu (modifiye PCWG2'ye g�re) veya BT veya MRG ile yumu�ak doku lezyonlar�ndaki progresyonu (RECIST 1.1'e g�re) kaps�yordu.
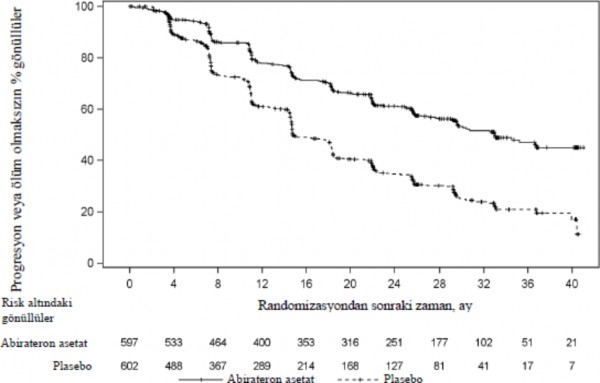
Tedavi gruplar� aras�nda rPFS bak�m�ndan anlaml� bir farkl�l�k g�zlendi (bkz. Tablo 1 ve �ekil 1).
Tablo 1: Radyografik Progresyonsuz Sa�kal�m - Katmanl� Analiz; Intent-to-treat Pop�lasyonu (�al��ma PCR3011) | ||
| AA-P | Plasebo |
Randomize hastalar | 597 | 602 |
Olay | 239 (%40,0) | 354 (%58,8) |
Sans�rlendi | 358 (%60,0) | 248 (%41,2) |
Olaya Kadar Ge�en S�re (ay) |
|
|
Medyan (%95 GA) | 33,02 (29,57, NE) | 14,78 (14,69, 18,27) |
Aral�k | (0,0+, 41,0+) | (0,0+, 40,6+) |
p de�eria | < 0,0001 |
|
Risk oran� (%95 GA)b | 0,466 (0,394, 0,550) |
|
Not: += sans�rlenmi� g�zlem, NE=hesaplanamaz. rPFS olay�n� tan�mlamada radyografik progresyon ve �l�m dikkate al�nm��t�r. AA-P= Abirateron asetat ve prednizolon alan hastalar. a p de�eri, ECOG PS skoruna (0/1 veya 2) ve visseral varl���na (yok veya var) g�re
tabakaland�r�lm�� bir log-rank testinden hesaplanm��t�r.
b Tehlike oran� tabakaland�r�lm�� orant�sal riskler modelinden hesaplanm��t�r. Tehlike oran�
< 1 AA- P lehine
![]()
�ekil 1: Radyografik Progresyonsuz Sa�kal�ma ili�kin Kaplan-Meier Grafi�i; Intent-to- treat Pop�lasyonu (�al��ma PCR3011)
![]()
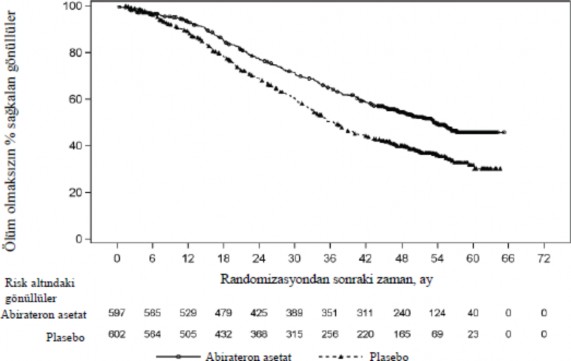
OS bak�m�ndan Plasebo art� ADT (TO=0,66; %95 GA: 0,56, 0,78; p<0,0001) ile kar��la�t�r�ld���nda, �l�m riskinde %34'l�k bir azalma ile AA-P art� ADT lehine istatistiksel olarak anlaml� bir iyile�me g�zlenmi�tir (bkz. Tablo 2 ve �ekil 2).
Tablo 2: PCR3011 �al��mas�nda Abirateron asetat veya Plasebo ile Tedavi Edilen Hastalar�n Genel Sa�kal�m� (Intent-to-treat Analiz) | ||
Genel Sa�kal�m | Abirateron asetat ve Prednizon | Plasebo (N=602) |
�l�m (%) | 275 (% 46) | 343 (% 57) |
Medyan sa�kal�m (aylar) | 53,3 | 36,5 |
(% 95 GA) | 48,2, NE | 33,5, 40,0 |
Tehlike oran� (% 95 GA)1 | 0,66 (0,56, 0,78) | |
NE=Hesaplanmad�
Tehlike oran� tabakaland�r�lm�� orant�sal riskler modelinden hesaplanm��t�r. Tehlike oran� <1 Abirateron asetat ve prednizon lehine
![]()
�ekil 2: Genel Sa�kal�ma ili�kin Kaplan-Meier Grafi�i; Intent-to-treat Pop�lasyon
Analizi (�al��ma PCR3011)
Alt grup analizleri abirateron asetat ile tedaviyi s�rekli olarak desteklemektedir. �nceden belirlenmi� alt gruplarda AA-P'nin rPFS ve OS �zerindeki tedavi etkisi genel �al��ma genel �al��ma pop�lasyonunda daha �st�n ve tutarl� olurken, ECOG skoru 2 olan alt grupta lehde bir yarar g�zlenmemi�tir, ancak �rneklem b�y�kl���n�n k���k olmas� (n=40) anlaml� bir sonu� ��kar�lmas�n� k�s�tlam��t�r.
![]()
Genel sa�kal�m ve rPFS'de g�zlenen art��lara ilave olarak, prospektif olarak tan�mlanan t�m ikincil sonlan�m noktalar� i�in plasebo kar��s�nda BYRETU lehine a�a��daki faydalar g�sterilmi�tir:.
�al��ma 302 (daha �nce kemoterapi almam�� hastalar)
Bu �al��maya asemptomatik veya hafif d�zeyde semptomatik olan ve hen�z kemoterapi endikasyonu bulunmayan kemoterapi almam�� hastalar dahil edilmi�tir. K�sa A�r� Envanteri - K�sa Formunun (BPI-00SF) “son 24 saat i�indeki en k�t� a�r�” maddesinin puan�n�n 0-1 olmas� asemptomatik, puan�n 2-3 olmas� ise hafif semptomatik olarak de�erlendirildi.
�al��ma 302'de (n = 1.088) yer alan hastalar�n medyan ya�� abirateron asetat ile birlikte prednizon veya prednizolon alan hastalar i�in 71, plasebo ile birlikte prednizolon alan hastalar i�in 70 idi. Abirateron asetat ile tedavi edilen hastalar�n �rklar�na g�re da��l�m� ��yleydi: 520 (%95,4) beyaz �rk, 15 (%2,8) siyah �rk, 4 (%0,7) sar� �rk ve 6 (%1,1) di�er �rklar. Do�u Ortak Onkoloji Grubu (ECOG) performans durumu, her iki koldaki hastalar�n %76's� i�in 0 ve
%24'� i�in 1 idi. Hastalar�n %50'sinde yaln�zca kemik metastazlar�, %31'inde kemik ve
yumu�ak doku veya lenf nodumetastazlar�ve%19'undayaln�zca yumu�ak doku veya lenf nodu
metastazlar� meydana gelmi�tir. Visseral metastaz� olan hastalar �al��maya al�nmam��t�r. Ortak
birincil etkinlik sonlan�m noktalar�, genel sa�kal�m ve radyografik progresyonsuz sa�kal�m
(rPFS) idi. Ortak birincil sonlan�m noktalar� �l��mlerine ilave olarak; kanser a�r�s� i�in opiat kullan�m�na kadar ge�en s�re, sitotoksik kemoterapi ba�lang�c�na kadar ge�en s�re, ECOG performans durumunda ≥ 1 puanl�k k�t�le�meye kadar ge�en s�re ve Prostat Kanseri �al��ma Grubu 2 (PCWG2) kriterlerine g�re PSA progresyonuna kadar ge�en s�re kullan�larak tedavi faydas� da de�erlendirilmi�tir. Bariz klinik progresyon durumunda �al��ma tedavilerine son verilmi�tir. Ara�t�rmac� karar�yla, do�rulanm�� radyografik progresyon ile de tedavi kesilebilecekti.
Radyografik progresyonsuz sa�kal�m (rPFS) PCWG2 kriterlerinde (kemik lezyonlar� i�in) tan�mlanan seri g�r�nt�leme �al��malar�na ve Solid T�m�rlerde Yan�t De�erlendirme Kriterleri'ne (RECIST) g�re (yumu�ak doku lezyonlar� i�in) de�erlendirildi. rPFS analizinde, radyografik progresyon de�erlendirmesi merkezi inceleme ile yap�lm��t�r.
Planl� rPFS analizi 401 olay� kapsam��, abirateron asetat ile tedavi edilen hastalar�n 150'sinde (%28) ve plasebo ile tedavi edilen hastalar�n 251'inde (%46) ya radyografik progresyon bulgusu g�r�lm�� ya da hastalar �lm��t�r. Tedavi gruplar� aras�nda rPFS bak�m�ndan anlaml� bir farkl�l�k g�zlenmi�tir (bkz. Tablo 3 ve �ekil 3).
Tablo 3: �al��ma 302: Abirateron Asetat veya plasebo ile birlikte prednizolon ve LHRH analoglar� ya da �ncesinde or�iektomi olan hastalardaki radyolografik progresyonsuz sa�kal�m | ||
| Abirateron Asetat (N = 546) | Plasebo (N = 542) |
Radyografik Progresyonsuz Sa�kal�m (rPFS) |
|
|
Progresyon veya �l�m | 150 (%28) | 251 (%46) |
Medyan rPFS (ay) (%95 GA) | Ula��lmad� (11,66; NE) | 8,3 (8,12; 8,54) |
p-de�eri* | < 0.0001 | |
Risk oran�** (%95 GA) | 0,425 (0,347; 0,522) | |
NE = Hesaplanmad�
* p-de�eri, ba�lang��taki ECOG skoruna (0 veya 1) g�re tabakaland�r�lm�� bir log-rank testinden hesaplanm��t�r.
** Risk oran� < 1 Abirateron asetat lehine
�ekil 3: Kaplan Meier grafi�inde Abirateron asetat veya plasebo ile birlikte prednizolon ve LHRH analoglar� ya da �ncesinde or�iektomi olan hastalardaki radyografik progresyonsuz sa�kal�m
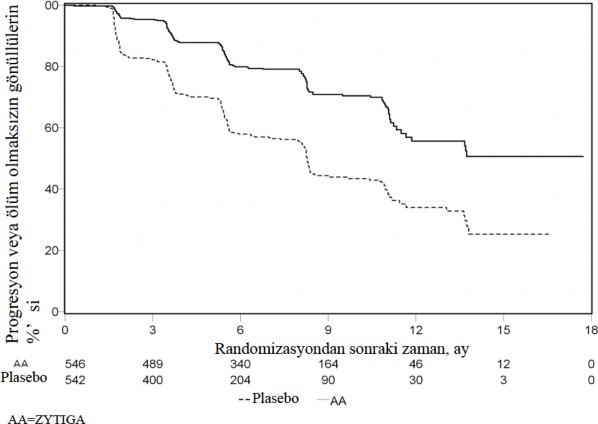
Ancak, Genel Sa�kal�ma (OS) ili�kin ikinci ara analiz tarihine kadar hasta verisi toplanmaya devam edilmi�tir. Ara�t�rmac�n�n rPFS �zerinde bir takip duyarl�l�k analizi olarak yapt��� radyografik inceleme Tablo 4 ve �ekil 4'te sunulmu�tur.
271'i (%50) abirateron asetat grubundan, 336's� (%62) plasebo grubundan olmak �zere toplam 607 hasta radyografik progresyon g�stermi� veya �lm��t�r. Abirateron asetat ile tedavi, plaseboya k�yasla radyografik progresyon riskini %47'ye kadar azaltm��t�r (TO = 0,530; 95% GA: [0,451; 0,623], p < 0,0001). Medyan rPFS, abirateron asetat grubunda 16,5 ay, plasebo grubunda ise 8,3 ay olmu�tur.
Tablo 4: �al��ma 302 Abirateron asetat veya plasebo ile birlikte prednizolon ve LHRH analoglar� ya da �ncesinde or�iektomi olmu� hastalardaki radyografik progresyonsuz sa�kal�m (�kinci ara analizdeki OS- Ara�t�r�c� �ncelemesi) | ||
| Abirateron asetat (N = 546) | Plasebo (N = 542) |
Radyografik Progresyonsuz Sa�kal�m (rPFS) |
|
|
Progresyon veya �l�m | 271 (%50) | 336 (%62) |
Medyan rPFS (ay) (%95 GA) | 16,5 (13,80; 16,79) | 8,3 (8,05; 9,43) |
p-de�eri* | < 0,0001 | |
Risk oran�** (%95 GA) |
0,530 (0,451; 0.623) | |
* p-de�eri, ba�lang��taki ECOG skoruna (0 veya 1) g�re tabakaland�r�lm�� bir log-rank testinden hesaplanm��t�r.
** Risk oran� < 1 Abirateron asetat lehine
�ekil 4: Kaplan Meier grafi�inde Abirateron asetat veya plasebo ile birlikte prednizolon ve LHRH analoglar� ya da �ncesinde or�iektomi olmu� hastalardaki radyografik progresyonsuz sa�kal�m (�kinci ara analizdeki OS- Ara�t�r�c� �ncelemesi)
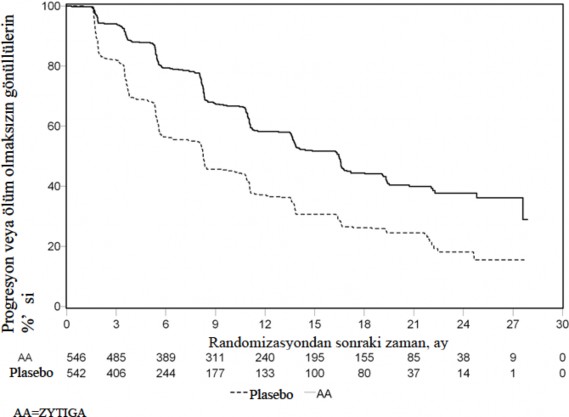
333 �l�m g�zlendikten sonra, OS i�in planl� bir ara analiz (IA) yap�ld�. G�zlenen klinik faydan�n b�y�kl��� dikkate al�narak �al��man�n k�rl��� kald�r�ld� ve plasebo grubundaki hastalara abirateron asetat ile tedavi olmalar� teklif edildi. Abirateron asetat �l�m riskini %25 azaltarak plaseboya k�yasla daha uzun bir genel sa�kal�m sa�lad� (TO = 0,752; %95 GA: [0,606; 0,934], p = 0,0097), ancak OS yeterli d�zeye eri�memi�ti ve ara analiz sonu�lar� istatistiksel anlaml�l�k i�in �nceden belirlenmi� sonland�rma s�n�r�n� kar��lam�yordu (bkz. Tablo 3). Bu ara analiz sonras�nda, sa�kal�m takibine devam edildi.
OS i�in planlanan nihai analiz 741 �l�m g�zlendikten sonra yap�ld� (medyan takip s�resi 49 ay). Abirateron asetat ile tedavi edilen hastalar�n %65'inin (354/546), plasebo ile tedavi edilen hastalar�n ise %71'inin (387/542) �ld��� saptand�. �l�m riskinde %19,4'l�k bir azalma ile, OS bak�m�ndan BYRETU ile tedavi edilen grup lehine istatistiksel olarak anlaml� bir fayda (TO = 0,806; %95 GA: [0,697; 0,931], p = 0,0033) ve 4,4 ayl�k medyan OS art��� sa�lanm��t�r (abirateron asetat 34,7 ay, plasebo 30,3 ay) (bkz. Tablo 5 ve �ekil 5). Bu iyile�me, plasebo kolundaki hastalar�n %44'� m�teakip tedavi olarak abirateron asetat alm�� olmalar�na ra�men g�sterilmi�tir.
Tablo 5: �al��ma 302: Abirateron asetat veya plasebo ile birlikte prednizolon ve LHRH analoglar� ya da �ncesinde or�iektomi olmu� hastalardaki genel sa�kal�m | ||
| Abirateron asetat (N = 546) | Plasebo (N = 542) |
Ara sa�kal�m analizi |
|
|
�l�m vakalar� (%) | 147 (%27) | 186 (%34) |
Ortalama sa�kal�m (ay) (%95 GA) | Ula��lmad� (NE; NE) | 27.2 (25,95; NE) |
p-de�eri* | 0.0097 | |
Risk oran�** (%95 GA) | 0.752 (0.606; 0.934) | |
Nihai sa�kal�m analizi |
| |
�l�m Vakalar� | 354 (%65) | 387 (%71) |
Medyan genel sa�kal�m (ay, %95 GA)] | 34,7 (32,7; 36,8) | 30,3 (28,7; 33,3) |
p-de�eri* | 0,0033 | |
Risk oran�** (%95 GA) | 0,806 (0,697; 0,931) | |
NE = Hesaplanmad�
* p-de�eri, ba�lang��taki ECOG skoruna (0 veya 1) g�re tabakaland�r�lm�� bir log-rank testinden hesaplanm��t�r.
** Risk oran� < 1 Abirateron asetat lehine
�ekil 5: Kaplan Meier grafi�inde Abirateron asetat veya plasebo ile birlikte prednizolon ve LHRH analoglar� ya da �ncesinde or�iektomi olmu� hastalardaki genel sa�kal�m - nihai analiz
Genel sa�kal�m ve rPFS'de g�zlenen art��lara ilave olarak, t�m sekonder sonlan�m noktalar� i�in plasebo kar��s�nda abirateron asetat lehine a�a��daki faydalar g�sterilmi�tir:
PCWG2 kriterlerine g�re PSA progresyonuna kadar ge�en s�re: PSA progresyonuna kadar ge�en medyan s�re abirateron asetat alan hastalar i�in 11,1 ay, plasebo alan hastalar i�in ise 5,6 ay olmu�tur (TO = 0,488; %95 GA: [0,420; 0,568], p < 0,0001). PSA progresyonuna kadar ge�en s�re abirateron asetat tedavisi ile yakla��k iki kat�na ��km��t�r (TO = 0,488). PSA yan�t� belgelenmi� olan hastalar�n oran� abirateron asetat grubunda, plasebo grubuna oranla daha fazla olmu�tur (%24 kar��s�nda %62, p=0,0001). Yumu�ak doku hastal��� �l��lebilir olan hastalarda, abirateron asetat ile anlaml� d�zeyde artm�� tam ve k�sm� yan�t oranlar� g�r�lm��t�r.
Kanser a�r�s� i�in opiat kullan�m�na kadar ge�en s�re: Nihai analiz s�ras�nda, prostat kanseri a�r�s� i�in opiat kullan�m�na kadar ge�en medyan s�re abirateron asetat alan hastalar i�in 33,4 ay, plasebo alan hastalar i�in 23,4 ay olmu�tur; (TO = 0,721; %95 GA: [0,614; 0,846], p < 0,0001).
Sitotoksik kemoterapi ba�lang�c�na kadar ge�en s�re: Sitotoksik kemoterapi ba�lang�c�na kadar ge�en medyan s�re abirateron asetat alan hastalar i�in 25,2 ay, plasebo alan hastalar i�in 16,8 ay olmu�tur (TO = 0,580; %95 GA: [0,487; 0,691], p < 0,0001).
ECOG performans skorunda ≥ 1 puan gerilemeye kadar ge�en s�re: ECOG performans skorunda
≥ 1 puan gerilemeye kadar ge�enmedyans�reabirateronasetat alan hastalar i�in 12,3 ay, plasebo
A�a��daki �al��ma sonlan�m noktalar� abirateron asetat tedavisi lehine istatistiksel olarak anlaml� bir �st�nl�k g�stermi�tir:
Objektif yan�t: Objektif yan�t, RECIST kriterlerine g�re �l��lebilir hastal��a sahip olup tam veya k�smi yan�t elde eden hastalar�n oran� olarak tan�mlanm��t�r (hedef lezyon olarak de�erlendirilebilmesi i�in ba�lang��taki lenf nodu b�y�kl���n�n ≥ 2 cm olmas� gereklidir). Ba�lang��ta �l��lebilir hastal��� olup objektif yan�t elde eden hastalar�n oran� abirateron asetat grubunda %36, plasebo grubunda ise %16 olmu�tur (p < 0,0001).
A�r�: Abirateron asetat tedavisi, plasebo ile kar��la�t�r�ld���nda, ortalama a�r� �iddeti progresyonunu anlaml� �ekilde %18'e kadar azaltm��t�r (p = 0,0490). Progresyona kadar ge�en medyan s�re abirateron asetat grubunda 26,7 ay, plasebo grubunda ise 18,4 ay olmu�tur.
FACT-P (Toplam Skor) anketinde k�t�le�meye kadar ge�en s�re: Abirateron asetat tedavisi, plasebo ile kar��la�t�r�ld���nda, FACT-P (Toplam Skor) anketindeki k�t�le�me riskini anlaml� �ekilde
%22'ye kadar azaltm��t�r (p = 0,0028). FACT-P (Toplam Skor) anketinde k�t�le�meye kadar ge�en s�re abirateron asetat grubunda 12,7 ay, plasebo grubunda ise 8,3 ay olmu�tur.
�al��ma 301 (daha �nce kemoterapi alm�� olan hastalar)
�al��ma 301'e daha �nceden dosetaksel kullanm�� olan hastalar dahil edilmi�tir. Bu kemoterapiden kaynaklanan toksisite tedavinin kesilmesine neden olmu� olabilece�i i�in, hastalar�n dosetaksel tedavisi s�ras�nda hastal�k ilerlemesi g�stermi� olmas� gerekmemi�tir. Hastalar protokole tan�mlanan radyografik ilerleme ve semptomatik ya da klinik ilerleme ile birlikte PSA ilerlemesi oluncaya kadar (hastan�n ba�lang��/en d���k d�zeyine g�re do�rulanm�� %25 art��) �al��ma tedavilerine devam etmi�tir. Daha �nceden prostat kanseri i�in ketokonazol tedavisi g�ren hastalar bu �al��maya al�nmam��t�r. Birincil etkililik sonlan�m noktas� genel sa�kal�m olmu�tur.
�al��maya al�nan hastalar�n medyan ya�� 69 idi (ya� aral��� 39 - 95 ya�). Abirateron asetat ile tedavi edilen hastalar�n �rklar�na g�re da��l�m� ��yleydi: 737'si (%93,2) beyaz �rktan, 28'i (%3,5) siyah �rktan, 11'i (%1,4) sar� �rktan ve geri kalan 14'� (%1,8) ise di�er �rklardand�. �al��maya al�nan hastalar�n %11'inin ECOG performans skoru 2 idi; %70'i i�in PSA ilerlemesi olsun ya da olmas�n, hastal�k ilerlemesine dair radyografik kan�tlar mevcuttu; %70'i daha �nce bir sitotoksik kemoterapi,
%30'u ise iki sitotoksik kemoterapi alm��t�. Abirateron asetat ile tedavi edilen hastalar�n %11'inde karaci�er metastaz� vard�.
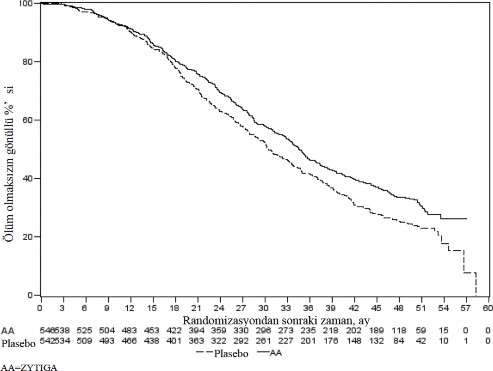
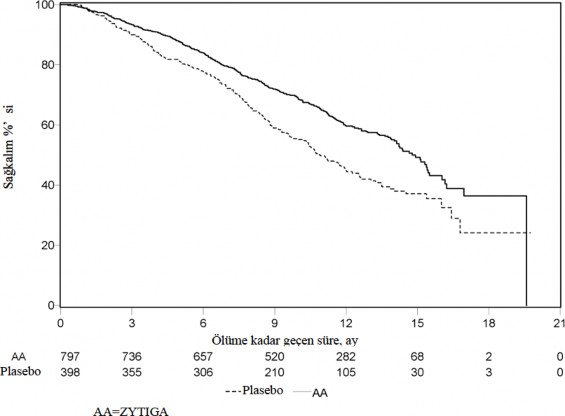
552 �l�m g�zlendikten sonra yap�lan planl� bir analizde, abirateron asetat ile tedavi edilen hastalar�n
%42'sinin (797 hastan�n 333'�), plasebo ile tedavi edilen hastalar�n ise %55'inin (398 hastan�n 219'u) �ld��� saptand�. Abirateron asetat ile tedavi edilen hastalar�n medyan genel sa�kal�mda istatistiksel olarak anlaml� bir art�� g�r�ld� (bkz. Tablo 6).
Tablo 6: Abirateron asetat veya plasebo ile birlikte prednizolon ve LHRH analoglar� ya da �ncesinde or�iektomi olmu� hastalardaki genel sa�kal�m | ||
| Abirateron asetat (N=797) | Plasebo (N=398) |
Birincil Sa�kal�m Analizi |
|
|
�l�mler (%) | 333 (%42) | 219 (%55) |
Medyan genel sa�kal�m (ay) (%95 GA) | 14,8 (14,1; 15,4) | 10,9 (10,2; 12,0) |
p-de�eria | < 0,0001 | |
Risk oran� (%95 GA) b | 0,646 (0,543; 0,768) | |
G�ncel Sa�kal�m Analizi |
|
|
�l�mler (%) | 501 (%63) | 274 (%69) |
Medyan genel sa�kal�m (ay) (%95 GA) | 15,8 (14,8; 17,0) | 11,2 (10,4; 13,1) |
Risk oran� (%95 GA) b | 0,740 (0,638; 0,859) | |
a p-de�eri ECOG performans durumu skoruna (0-1 veya 2), a�r� skoruna (yok veya var), daha �nceden al�nm�� kemoterapi rejimlerinin say�s�na (1 veya 2) ve hastal�k ilerlemesinin tipine (yaln�zca PSA veya radyografik) g�re tabakala�t�r�lm�� bir logaritmik-s�ralama testinden elde edilmi�tir.
b Tehlike oran� tabakaland�r�lm�� orant�sal bir risk modeline g�re hesaplanm��t�r. Tehlike oran� < 1 abirateron asetat lehine
Tedavinin ba�lang��taki ilk birka� ay�ndan sonra her de�erlendirme noktas�nda, plasebo ile tedavi edilip hayatta kalan hastalar�n oran�yla kar��la�t�r�ld���nda, abirateron asetat ile tedavi edilen hastalar�n daha y�ksek sa�kal�m oran�na sahip oldu�u belirlenmi�tir (bkz. �ekil 6).
�ekil 6: Kaplan Meier grafi�inde Abirateron asetat veya plasebo ile birlikte prednizolon ve LHRH analoglar� ya da �ncesinde or�iektomi olmu� hastalardaki genel sa�kal�m
Altgrup sa�kal�m analizleri Abirateron asetat ile tedavisi i�in tutarl� bir sa�kal�m faydas� g�sterdi (bkz. �ekil 7).
�ekil 7: Alt gruplara g�re genel sa�kal�m: risk oran� ve %95'lik g�ven aral���
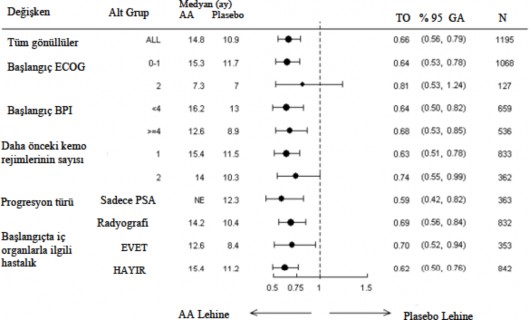
AA= Abirateron asetat; BPI=K�sa a�r� envanteri; GA=G�ven aral���; ECOG=do�u kooperatif onkoloji grubu (eastern cooperative oncology grup) performans skoru; TO=Tehlike oran�; NE= Hesaplanmad�
Genel sa�kal�mda g�zlenen art��a ek olarak, t�m ikincil sonlan�m noktalar� abirateron asetat lehine idi ve a�a��da g�sterildi�i �ekilde bu farkl�l�klar birden fazla test i�in ayarland���nda istatistiksel olarak anlaml� idi:
Abirateron asetat alan hastalarda, plasebo alanlara g�re istatistiksel olarak anlaml� d�zeylerde daha y�ksek PSA yan�t oran� (ba�lang�� de�erinden ≥%50 olarak tan�mlanm��) elde edildi, abirateron asetat ile %38 iken, plaseboyla %10, p < 0,0001.
Abirateron asetat ile tedavi edilen hastalarda, PSA ilerlemesine kadar olan medyan s�re 10,2 ay iken, bu s�re plasebo ile tedavi edilen hastalarda 6,6 ay idi (TO=0,580; %95 GA: [0,462; 0,728], p < 0,0001).
A�r�
A�r�s�nda hafifleme olan hastalar�n oran� abirateron asetat grubunda, plasebo grubuna oranla istatistiksel olarak anlaml� derecede daha fazlayd� (%44'e kar�� %27, p=0,0002). Bir hastada d�rt hafta arayla yap�lan iki ard���k de�erlendirmede 24 saatlik bir s�rede analjezik kullan�m skorunda herhangi bir art�� olmaks�z�n BPI-SF en k�t� a�r� yo�unlu�u skorunda ba�lang�ca g�re en az %30 azalma sa�lanmas�, a�r�n�n hafiflemesi yan�t� olarak tan�mland�. A�r� palyasyonu i�in yaln�zca ba�lang�� a�r� puan� ≥ 4 olan hastalar de�erlendirildi ve en az bir ba�lang��-sonras� a�r� skoru analiz edildi (N=512).
Abirateron asetat ile tedavi edilen hastalarda, plasebo ile tedavi edilenlere oranla daha d���k oranda a�r� art��� g�r�ld�: 6. ayda abirateron asetat tedavisi alanlar�n %22'sine kar��l�k plasebo alanlar�n %28'i, 12. ayda abirateron asetat tedavisi alanlar�n %30'una kar��l�k plasebo alanlar�n
%38'i ve 18. ayda abirateron asetat tedavisi alanlar�n %35'ine kar��l�k plasebo alanlar�n %46's�nda a�r�da art�� vard�. A�r� art��� boyunca, iki ard���k de�erlendirmede son 24 saatlik bir s�rede analjezik kullan�m skorunda herhangi bir azalma olmaks�z�n BPI-SF en k�t� a�r� yo�unlu�u skorunda ba�lang�ca g�re en az %30 veya daha fazla bir art�� ya da iki ard���k de�erlendirmede analjezik kullan�m skorunda %30 veya daha fazla bir art�� olarak tan�mland�. 25. persentilde a�r� ilerlemesine kadar ge�en s�re abirateron asetat grubunda 7,4 ay iken, plasebo grubunda 4,7 ay idi.
�skelet sistemiyle ili�kili olaylar
5.2. Farmakokinetik �zellikler
Genel �zelliklerAbirateron asetat uygulamas�ndan sonra, abirateron farmakokineti�i sa�l�kl� g�n�ll�lerde,
ilerlemi� metastatik prostat kanserli hastalarda ve kanser hastas� olmayan karaci�er veya b�brek yetmezli�i olan g�n�ll�lerde �al���lm��t�r. Abirateron asetat in vivo olarak h�zla bir androjen biyosentez inhibit�r� olan abiraterona d�n���r (bkz. B�l�m 5.1).
Emilim:
Abirateron asetat a�l�k durumunda oral yoldan uyguland�ktan sonra, em y�ksek abirateron konsantrasyonlar�na yakla��k 2 saatte ula��l�r.
Abirateron asetat�n yemekle birlikte al�nmas�, yeme�in ya� i�eri�ine ba�l� olarak, a� kar�na al�nmas�na oranla 10 kata kadar (EAA a��s�ndan) ve 17 kata kadar (Cmaks a��s�ndan) daha fazla ortalama sistemik abirateron maruziyetine yol a�ar. Yemeklerin i�erik ve bile�imindeki normal farkl�l�klar g�z �n�ne al�nd���nda, BYRETU'nun yemeklerle birlikte al�nmas� olduk�a de�i�ken maruziyetler ile sonu�lanma potansiyeline sahiptir. BYRETU tabletler a� karn�na g�nde bir kez tek doz olarak al�nmal�d�r. BYRETU, yemekten en az iki saat sonra al�nmal�d�r ve BYRETU al�nd�ktan sonra en az bir saat yemek yenmemelidir. Tabletler b�t�n olarak suyla yutulmal�d�r (bkz. B�l�m 4.2).
Da��l�m:
�nsan plazmas�nda 14C-abirateronun proteine ba�lanma oran� %99,8'dir. G�r�n�r da��l�m hacmi yakla��k 5.630 litre olup, abirateronun periferik dokulara yo�un bir �ekilde da��ld���n� g�sterir.
Biyotransformasyon:
C-abirateron asetat kaps�l formunda oral yoldan al�nd�ktan sonra, abirateron asetat, abiraterona
hidrolize olur ve daha sonra birincil olarak karaci�erde olmak �zere s�lfasyon, hidroksilasyon ve oksidasyona u�rar. Dola��mdaki radyoaktivitenin b�y�k �o�unlu�u (yakla��k %92) abirateronun metabolitleri halinde bulunur. Belirlenebilen 15 metabolitten iki temel metabolit, her biri toplam radyoaktivitenin yakla��k %43'�n� temsil eden abirateron s�lfat ve N-oksit abirateron s�lfatt�r.
Eliminasyon:
Sa�l�kl� g�n�ll�lerden elde edilen verilere g�re, plazmadaki abirateronun yar�lanma s�resi yakla��k 15 saattir. 1.000 mg 14C-abirateron asetat�n oral yoldan al�nmas�ndan sonra radyoaktif dozun yakla��k %88'i fe�este ve yakla��k %5'i idrarda g�r�l�r. Fe�este bulunan ana bile�ikler de�i�memi� abirateron asetat ve abiraterondur (s�ras�yla uygulanan dozun yakla��k %55 ve %22'si).
Hastalardaki karekteristik �zellikler
Karaci�er yetmezli�i olan hastalar:
Abirateron asetat�n farmakokineti�i hafif ya da orta �iddette karaci�er yetmezli�i olan hastalar (s�ras�yla Child-Pugh S�n�f A ve B) ile sa�l�kl� kontrollerde �al���lm��t�r. 1.000 mg'l�k tek bir oral doz sonras� abiraterona sistemik maruziyet hafif ve orta �iddette karaci�er yetmezli�i olan hastalarda s�ras�yla %11 ve %260 oran�nda artmaktad�r. Abirateronun ortalama yar�lanma s�resi hafif karaci�er yetmezli�i olan hastalarda yakla��k 18 saate ve orta �iddette karaci�er yetmezli�i olan hastalarda ortalama 19 saate uzamaktad�r.
Bir ba�ka �al��mada, abirateronun farmakokineti�i daha �nceden ciddi karaci�er yetmezli�i (Child-Pugh S�n�f C) olan (n=8) ve normal hepatik fonksiyonu olan 8 sa�l�kl� birey �zerinde incelenmi�tir. Abirateronenin sistemik etkisi (EAA), yakla��k %600 oran�nda art�� g�stermi� ve serbest ilac�n etkisi �iddetli karaci�er bozuklu�u olanlarda normal karaci�er fonksiyonu olanlara g�re %80 oran�nda artm��t�r.
Daha �nceden hafif karaci�er yetmezli�i olan hastalarda doz ayarlamas�na gerek yoktur. BYRETU kullan�m� faydan�n olas� riskten a��k�a a��r bast���, orta �iddette karaci�er yetmezli�i olan hastalarda dikkatle de�erlendirilmelidir (bkz. B�l�m 4.2 ve 4.4). BYRETU a��r karaci�er yetmezli�i olan hastalarda kullan�lmamal�d�r (bkz. B�l�m 4.2, 4.3 ve 4.4).
BYRETU tedavisi s�ras�nda hepatotoksisite geli�en hastalarda, tedavinin durdurulmas� ve doz ayarlamas� gerekebilir (bkz. B�l�m 4.2 ve 4.4).
B�brek yetmezli�i olan hastalar:
Stabil bir hemodiyaliz program�nda olan son d�nem b�brek yetmezli�i olan hastalar ile b�brek fonksiyonlar� normal olan e�lenmi� kontrol hastalar�nda Abirateron asetat�n farmakokineti�i kar��la�t�r�lm��t�r. Diyaliz program�nda olan son d�nem b�brek yetmezli�i olan hastalarda 1.000 mg'l�k tek bir oral doz sonras� abiraterona sistemik maruziyette art�� olmam��t�r. A��r b�brek yetmezli�i dahil, b�brek yetmezli�i olan hastalarda Abirateron asetat�n uygulanmas� s�ras�nda dozu azaltmaya gerek yoktur (bkz. B�l�m 4.2). Ancak prostat kanseri ve a��r b�brek yetmezli�i olan hastalarla ilgili klinik deneyim bulunmamaktad�r. Bu t�r hastalarda dikkatli olunmas� tavsiye edilir.
5.3. Klinik �ncesi g�venlilik verileri
T�m hayvan toksisite �al��malar�nda, dola��mdaki testesteron d�zeyleri anlaml� derecelerde azalm��t�r. Buna ba�l� olarak �reme organlar�yla adrenal, hipofiz ve meme bezlerinin a��rl�klar�nda azalma ile morfolojik ve/veya histopatolojik de�i�iklikler g�zlenmi�tir. T�m de�i�iklikler tamamen ya da k�smen geri d�nd�r�lebilir nitelikteydi. �reme organlar� ve androjene duyarl� organlardaki de�i�iklikler abirateronun farmakolojisiyle uyumludur. Tedaviyle ili�kili t�m hormonal de�i�iklikler eski haline d�nm��t�r veya 4 haftal�k bir toparlanma d�nemi sonunda geri d�zeldi�i g�sterilmi�tir.
Erkek ve di�i s��anlarda yap�lan fertilite �al��malar�nda, abirateron asetat fertiliteyi azaltm��, fakat abirateron asetat kesildikten sonra 4 ila 16 haftada fertilite tam olarak eski haline d�nm��t�r.
S��anlarda yap�lan bir geli�im toksisitesi �al��mas�nda, abirateron asetat azalm�� fet�s a��rl���nda
ve sa�kal�m da dahil olmak �zere gebeli�i etkilemi�tir. Abirateron asetat teratojenik olmasa da, d�� genital organlarda etkiler g�r�lm��t�r.
S��anlarda y�r�t�len bu fertilite ve geli�im toksisitesi �al��malar�nda, t�m etkiler abirateronun farmakolojik aktivitesine ba�l� olmu�tur.
T�m hayvan toksikoloji �al��malar�nda g�r�len �reme organlar�ndaki de�i�iklikler d���nda, klasik g�venlilik farmakolojisi, tekrarlayan doz toksisitesi, genotoksisite ve karsinojenik potansiyel �al��malar�ndan elde edilen klinik d��� veriler insanlar i�in �zel bir tehlikeyi g�stermemi�tir. Transjenik farelerde (Tg.rasH2) yap�lan 6 ayl�k bir �al��mada abirateron asetat karsinojenik bulunmam��t�r. S��anlarda yap�lan 24 ayl�k bir karsinojenisite �al��mas�nda, abirateron asetat testislerde interstisyel h�cre neoplazmalar�n�n insidans�n� artt�rm��t�r. Bu bulgu abirateronun farmakolojik etkisine ba�l� ve s��anlara �zg� olarak de�erlendirilmi�tir. Abirateron asetat di�i s��anlarda karsinojenik de�ildir.
�evresel Risk De�erlendirmesi
Abirateron etkin maddesi, akuatik ortam (�zellikle bal�klar) i�in �evresel bir risk olu�turur.
6. FARMAS�T�K �ZELL�KLER
6.1. Yard�mc� maddelerin listesi
Mikrokristalin sel�loz
Laktoz monohidrat (s���r s�t�nden elde edilir.) Povidon (K30)
Sodyum lauril s�lfat Kroskarmeloz sodyum Kolloidal silikon dioksit Magnezyum stearat
ayda, 12'nci ayda ve 18'inci ayda abirateron asetat grubunda iskelet sistemiyle ili�kili olay g�r�len hasta oran� plasebo ile kar��la�t�r�ld���nda daha d���kt� (s�ras�yla 6. ayda %18'e kar�� %28, 12. ayda %30'a kar�� %40 ve 18. ayda %35'e kar�� %40). 25. persentilde iskelet sistemiyle ili�kili ilk olay�n g�r�lme s�resi abirateron asetat grubunda 9,9 ay ile, 4,9 ay olan kontrol grubundan iki kat daha uzundu. Patolojik k�r�k, spinal kord bas�s�, kemi�e palyatif radyoterapi veya kemi�e y�nelik cerrahi giri�im, iskelet sistemiyle ili�kili olay olarak tan�mland�.
6. FARMAS�T�K �ZELL�KLER
6.1. Yard�mc� maddelerin listesi
Mikrokristalin sel�loz
Laktoz monohidrat (s���r s�t�nden elde edilir.) Povidon (K30)
Sodyum lauril s�lfat Kroskarmeloz sodyum Kolloidal silikon dioksit Magnezyum stearat
6.2. Ge�imsizlikler
Bilinen herhangi bir ge�imsizli�i bulunmamaktad�r.
6.3. Raf �mr�
24 ay
6.4. Saklamaya y�nelik �zel tedbirler
25 °C alt�ndaki oda s�cakl���nda saklanmal�d�r.
�ocuklar�n g�remeyece�i, eri�emeyece�i yerlerde ve ambalaj�nda saklay�n�z.
6.5. Ambalaj�n niteli�i ve i�eri�i
BYRETU, 120 tablet i�erecek �ekilde Alu - Alu folyo blister ile sunulmaktad�r.
6.6. Be�eri t�bbi �r�nden arta kalan maddelerin imhas� ve di�er �zel �nlemler
Etki mekanizmas�na dayanarak, bu t�bbi �r�n geli�mekte olan fet�se zarar verebilir, bu nedenle
gebe olan ya da gebe olma olas�l��� bulunan kad�nlar BYRETU ile korunmas�z temas etmemeli;
�rne�in eldiven kullanmal�d�r (bkz. B�l�m 4.6).
Kullan�lmam�� olan �r�nler ya da at�k materyaller “T�bbi At�klar�n Kontrol� Y�netmeli�i” ve “Ambalaj ve Ambalaj At�klar�n�n Kontrol� Y�netmelik”lerine uygun olarak imha edilmelidir. Bu t�bbi �r�n akuatik ortam i�in bir risk olu�turabilir (bkz. B�l�m 5.3).
 Deri Kanseri
Deri kanseri �ok rastlanan bir hastal�kt�r. �� ana t�r� bulunur ;genelde kemirici �lser olarak bilinen bazal h�creli karsinom, yass� h�creli karsinom ve k�t� huylu t�m�r.
Deri Kanseri
Deri kanseri �ok rastlanan bir hastal�kt�r. �� ana t�r� bulunur ;genelde kemirici �lser olarak bilinen bazal h�creli karsinom, yass� h�creli karsinom ve k�t� huylu t�m�r. |
 Omurilik zedelenmeleri
Omurilik zedelenmesini takip eden birka� g�n i�inde, hi�kimse hasarin ne kadar olacagini tahmin edemez. Buradaki sorun, omuriligin herhangi bir zedelenmesinden hemen sonra, bir omurilik sokunun olusmasidir.
Omurilik zedelenmeleri
Omurilik zedelenmesini takip eden birka� g�n i�inde, hi�kimse hasarin ne kadar olacagini tahmin edemez. Buradaki sorun, omuriligin herhangi bir zedelenmesinden hemen sonra, bir omurilik sokunun olusmasidir. |
�LA� E�DE�ERLER�
| E�de�er �la� Ad� | Barkodu | �la� Fiyat� |
|---|---|---|
| ABIRATEX | 8699541015349 | 19,856.26TL |
| ABITIGA | 8699638095513 | 18,760.27TL |
| ABYGA | 8699525019776 | 17,861.11TL |
| ARVILA | 8699586013331 | 17,866.96TL |
| BYRETU | 8699650012260 | 17,914.92TL |
| Di�er E�de�er �la�lar |
 |
Mide Kanseri Mide kanseri genellikle mideyi t�m�yle kaplayan ve mukus �retmekle g�revli h�crelerde ba�lar. Bu kanser tipine adenokarsinom denir. |
 |
Parkinson Hastal��� Hastal�k ilk kez 1817 de �ngiliz doktor James Parkinson taraf�ndan tan�mlanm�� ve Dr. Parkinson hastal��� “sallay�c� fel�” olarak kaleme alm��. |
 |
Kalp Krizi Kalbe giden kan ak��� durdu�unda kalp krizi meydana gelir. |
�LA� GENEL B�LG�LER�
Onko Ko�sel �la� San. Tic. A.�
| Sat�� Fiyat� | 17914.92 TL [ 26 Apr 2024 ] |
| �nceki Sat�� Fiyat� | 17914.92 TL [ 22 Apr 2024 ] |
| Original / Jenerik | Original �la� |
| Re�ete Durumu | Normal Re�eteli bir ila�d�r. |
| Barkodu | 8699650012260 |
| Etkin Madde | Abirateron |
| ATC Kodu | L02BX03 |
| Birim Miktar | 250 |
| Birim Cinsi | MG |
| Ambalaj Miktar� | 120 |
| Antineoplastik ve �mm�nomod�lat�r Ajanlar > Hormon Antagonistleri > Abirateron |
| Yerli ve Be�eri bir ila�d�r. |