VIDAZA 100 mg SC enjeksiyonluk s�spansiyon i�in toz i�eren 1 flakon K�sa �r�n Bilgisi
{ Azacitidine }
1. BE�ER� TIBB� �R�N�N ADI
VIDAZA® 100 mg SC enjeksiyonluk s�spansiyon i�in toz i�eren flakon Steril
Sitotoksik
2. KAL�TAT�F VE KANT�TAT�F B�LE��M
Etkin madde
Her bir flakon 100 mg azasitidin i�erir.Haz�rlama sonras� elde edilen s�spansiyon her mL'de 25 mg azasitidin i�erir.
Yard�mc� maddeler
Yard�mc� maddeler i�in 6.1'e bak�n�z.
3. FARMAS�T�K FORMU
Enjeksiyonluk s�spansiyon i�in toz. Beyaz liyofilize toz.
4 mL enjeksiyonluk su i�eren ��r�ngan�n i�nesi plastik kapakl� azasitidin flakonuna bat�r�lmal� ve enjeksiyonluk su flakona enjekte edilmelidir.
4. KL�N�K �ZELL�KLER
4.1. Terap�tik endikasyonlar
VIDAZA hematopoietik k�k h�cre transplantasyonuna uygun olmayan yeti�kin hastalarda:
Uluslararas� Prognostik Skorlama Sistemi'ne (IPSS) g�re intermediate 2 ve y�ksek risk miyelodisplastik sendrom (MDS)
4.2. Pozoloji ve uygulama �ekli
VIDAZA tedavisi kemoterap�tik ajanlar konusunda tecr�beli bir hekim taraf�ndan ba�lanmal� ve izlenmelidir. Hastalara, tedavi �ncesinde bulant� ve kusmaya kar�� anti-emetik premedikasyonu uygulanmal�d�r.
Pozoloji/uygulama s�kl��� ve s�resi:
�lk tedavi siklusunda tavsiye edilen ba�lang�� dozu tedavi �ncesi hematoloji laboratuvar de�erlerinden ba��ms�z olarak, t�m hastalar i�in v�cut y�zey alan�na g�re 75 mg/m2 dozunda olmal�, subkutan olarak 7 g�n boyunca yap�lan enjeksiyonlar� takiben 21 g�nl�k bir ara verilmelidir (28 g�n s�ren tedavi siklusu).
Hastalar�n en az 6 siklus tedavi almas� �nerilir. Hasta tedaviden fayda g�rd��� s�rece ya da hastal�kta ilerleme g�r�l�nceye kadar tedavi devam ettirilmelidir.
Hastalar hematolojik yan�t/toksisite ve renal toksisite a��s�ndan izlenmelidir (bak�n�z B�l�m 4.4); bir sonraki siklusa ba�larken erteleme ya da a�a��da belirtildi�i �ekilde doz azalt�m� gerekebilir.
VIDAZA ile oral azasitidin birbirinin yerine kullan�lmamal�d�r. Maruz kalmadaki farkl�l�klar nedeniyle, oral azasitidin i�in doz ve zamanlama �nerileri enjekte edilebilir azasitidin i�in olanlardan farkl�d�r. Sa�l�k profesyonellerinin t�bbi �r�n�n ad�n�, dozu ve uygulama yolunu do�rulamalar� �nerilir.
Laboratuvar Testleri:
Tedaviye ba�lamadan ve her tedavi siklusu �ncesinde; karaci�er fonksiyon testleri, serum kreatinin ve serum bikarbonat seviyeleri �l��lmelidir. Tedaviye ba�lamadan ve en az her tedavi siklusundan �nce, cevap ve toksisiteyi izlemek gerekli oldu�u i�in tam kan say�mlar� yap�lmal�d�r.
Hematolojik Toksisite Nedeniyle Doz Ayarlamas�:
Hematolojik toksisiteye ba�l� olarak doz ayarlamas�nda; Hematolojik toksisite, trombosit say�s� ≤ 50 x 109/L ve/veya mutlak n�trofil say�s� (MNS) ≤1 x 109/L ise, siklus i�erisinde ula��lan “en d���k de�er” (en d���k) olarak tan�mlan�r.
�yile�me ise hematolojik toksisite g�zlenen h�cre serilerinde ba�lang�� de�erleri ile en d���k de�er aras�ndaki mutlak fark�n en az yar�s� kadar bir art���n olma hali olarak tan�mlan�r. (iyile�me
≥ En d���k say�m + (0,5 x [|Ba�lang�� say�m – En d���k say�m|]).
Tedavi �ncesi ba�lang�ca g�re kan say�m� de�erleri d��memi� hastalarda (�rne�in beyaz kan h�cresi – BKH - ≥ 3 x 10 /L ve MNS ≥ 1,5 x 10 /L ve trombosit ≥ 75 x 10 /L) VIDAZA tedavisine ba�l� olarak hematolojik toksisite ortaya ��karsa, bir sonraki tedavi siklusu trombosit say�s� ve MNS de�erleri d�zelene kadar ertelenmelidir. 14 G�n i�erisinde de�erlerde iyile�me sa�lan�rsa herhangi bir doz de�i�ikli�ine gerek yoktur. Ancak 14 g�n i�erisinde iyile�me sa�lanamazsa bu durumda a�a��daki tabloya g�re doz azalt�lmas� yap�lmal�d�r. Doz ayarlamalar�n� takiben siklus 28 g�ne d�nd�r�lmelidir.
Siklustaki en d���k de�er | 14 g�n i�erisinde de�erlerde d�zelme olmazsa bir sonraki siklusda verilebilecek doz miktar� (%) | |
MNS (x 10 /l) | Trombosit (x 10 /l) | |
≤ 1 | ≤ 50 | %50 |
> 1 |
> 50 | %100 |
*�yile�me = Say�m ≥ En d���k say�m + (0,5 x [Ba�lang�� say�m – En d���k say�m])
Tedavi �ncesi ba�lang�ca g�re kan say�m� de�erleri d��m�� hastalarda (�rne�in beyaz kan h�cresi – BKH - < 3 x 10 /Lveya MNS < 1,5 x 10 /L veya trombosit < 75 x 10 /L) VIDAZA tedavisini takiben BKH ya da MNS ya da trombosit say�s�nda, uygulama �ncesine g�re ≤ %50 bir azalma ya da %50'den fazla olmas�na ra�men herhangi bir h�cre seri farkl�la�mas�nda iyile�me g�r�lmesi durumunda doz ayarlamas�na ya da tedavinin ertelenmesine gerek yoktur.
E�er BKH ya da MNS ya da trombosit say�s�ndaki azalma uygulama �ncesine g�re %50'den fazla ise ve herhangi bir h�cre seri farkl�la�mas�nda iyile�me g�r�lmemesi durumunda VIDAZA tedavisinin bir sonraki siklusu, trombosit say�s� ve MNS d�zelene kadar ertelenmelidir. 14 g�n i�erisinde iyile�me sa�lan�rsa herhangi bir doz ayarlamas�na gerek yoktur. Ancak 14 g�n s�resinde bir d�zelme g�zlenmemesi durumunda kemik ili�i h�cresel
yap�s� de�erlendirilmelidir. E�er kemik ili�i h�cre d�zeyi > %50 ise doz de�i�ikli�ine gerek yoktur. Kemik ili�i h�cre d�zeyi ≤%50 ise tedavi ertelenmeli ve doz a�a��daki tabloya g�re azalt�lmal�d�r.
Kemik �li�i H�cre D�zeyi | 14 g�n i�erisinde de�erlerde d�zelme izlenmezse yap�lmas� gereken doz de�i�ikli�i (%) | |
| �yile�me* ≤21 G�n | �yile�me* > 21 G�n |
%15-50 | %100 | %50 |
< %15 |
%100 |
%33 |
*�yile�me = Say�m ≥ en d���k say�m + (0,5 x [Ba�lang�� say�m – en d���k say�m]) Doz ayarlamalar�n� takiben siklus 28 g�ne d�nd�r�lmelidir.
Uygulama �ekli:
Suland�r�lm�� VIDAZA; �st kol b�lgesi, uyluk ya da kar�na subkutan olarak enjekte edilmelidir. Enjeksiyon yerleri d�n���ml� olarak de�i�tirilmelidir. Yeni enjeksiyonlar bir �ncekinden en az 2,5 cm uza�a yap�lmal� ve kesinlikle hassasiyet, ��r�k, k�zar�kl�k ya da sertle�me olan b�lgelere uygulanmamal�d�r. Suland�r�ld�ktan sonra s�spansiyon filtre edilmemelidir. VIDAZA i�in suland�rma ve uygulama prosed�r� i�in detayl� talimatlar b�l�m 6.6'da verilmi�tir.
�zel pop�lasyonlara ili�kin ek bilgiler:
B�brek Yetmezli�i:
Azasitidin, ba�lang�� doz ayarlamas� olmaks�z�n b�brek yetmezli�i olan hastalara uygulanabilir (bak�n�z B�l�m 5.2). E�er serum bikarbonat d�zeyinde nedeni a��klanamayan bir �ekilde 20 mmol/l'nin alt�nda azalma ortaya ��karsa, bir sonraki siklusta doz %50 azalt�lmal�d�r. E�er serum kreatinin ya da kan �re azot (BUN) de�erleri a��klanamayan bir �ekilde ba�lang�� de�erlerinin ≥ 2 kat �zerine ve normal de�erin en �st s�n�r� (ULN)'na ��karsa, de�erler normale ya da ba�lang�� d�zeylerine d�nene kadar bir sonraki siklus ertelenmeli ve takip eden tedavi siklusunda doz %50 azalt�lmal�d�r (bak�n�z B�l�m 4.4).
Karaci�er Yetmezli�i:
Karaci�er yetmezli�i olan hastalarda yap�lm�� �al��ma bulunmamaktad�r (bak�n�z B�l�m 4.4). Ciddi karaci�er yetmezli�i bulunan hastalar advers olaylar i�in dikkatlice izlenmelidir. Tedaviye ba�lamadan �nce karaci�er yetmezli�i olan hastalarda ba�lang�� dozu i�in spesifik bir doz de�i�ikli�i �nerilmemektedir; takip eden doz de�i�iklikleri hematolojik laboratuvar de�erleri �zerinden yap�lmal�d�r. �leri evre malign hepatik t�m�r� olan hastalarda VIDAZA kontrendikedir (bak�n�z B�l�m 4.3 ve 4.4).
Pediyatrik pop�lasyon:
Yeterli g�venlilik ve etkililik verisi bulunmad���ndan 18 ya� alt�ndaki �ocuklar ve ad�lesanlarda VIDAZA kullan�m� �nerilmemektedir. Halihaz�rda mevcut veriler b�l�m 4.8,
5.1 ve 5.2'de a��klanmaktad�r, ancak pozoloji konusunda herhangi bir tavsiyede bulunulmamaktad�r.
Geriyatrik pop�lasyon:
Ya�l� hastalar i�in spesifik bir doz ayarlamas� �nerilmemektedir. Ya�l�larda b�brek fonksiyonlar� zaten azald��� i�in b�brek fonksiyonlar�n�n izlenmesi yararl� olabilir.
4.3. Kontrendikasyonlar
Azasitidine veya b�l�m 6.1'de listelenen herhangi bir bile�enine a��r� duyarl��� olan hastalarda,
4.4. �zel kullan�m uyar�lar� ve �nlemleri
Hematolojik toksisite
Azasitidin ile tedavi esnas�nda, �zellikle ilk 2 siklus s�ras�nda (bkz. B�l�m 4.8), anemi, n�tropeni ve trombositopeni s�k g�zlenmektedir. Cevap ve toksisiteyi izlemek gerekli oldu�u i�in, en az her tedavi siklusundan �nce tam kan say�mlar� yap�lmal�d�r. �lk siklus i�in �nerilen dozun uygulanmas�ndan sonra, en d���k say�mlara ve hematolojik cevaba dayanarak (bkz. B�l�m 4.2), daha sonraki sikluslar i�in doz azalt�labilir veya uygulama geciktirilebilir.
Hastalara derhal febril ataklar�n� bildirmeleri tavsiye edilmelidir. Ayr�ca hastalara ve doktorlara kanama belirtileri ve semptomlar� i�in dikkatli olmalar� tavsiye edilir.
Karaci�er yetmezli�i
Karaci�er yetmezli�i olan hastalarda herhangi bir �al��ma yap�lmam��t�r. Metastatik hastal��a ba�l� olarak b�y�k t�m�r y�k� olan, �zellikle albumin alt s�n�r de�eri <30 g/L olan hastalarda, azasitidin tedavisi s�ras�nda ilerleyen karaci�er komas� ve �l�m seyrek olarak rapor edilmi�tir. Azasitidin, ilerlemi� malign karaci�er t�m�rleri olan hastalarda kontrendikedir (bkz. B�l�m 4.3).
B�brek yetmezli�i
Kemoterap�tik ajanlarla birlikte i.v. azasitidin ile tedavi edilen hastalarda serum kreatinin d�zeyi art���, b�brek yetmezli�i ve �l�mle sonu�lanan b�brek fonksiyon bozukluklar� bildirilmi�tir. Ek olarak, alkali idrar ve hipokalemi (serum potasyumu < 3mmol/L) ile birlikte serum bikarbonatlar�n�n <20 mmol/L'ye d��mesi olarak tan�mlanan renal t�b�ler asidoz, azasitidin ve etoposid ile tedavi edilen 5 kronik miyeloid l�semi (KML) hastas�nda geli�mi�tir. Serum kreatinin veya BUN seviyelerinde a��klanamayan art��lar veya serum bikarbonatta azalmalar (<20 mmol/L) olu�ur ise, dozaj azalt�lmal� veya uygulama geciktirilmelidir (bkz. B�l�m 4.2).
Hastalar, olig�ri ve an�ri durumunda derhal doktorlar�n� bilgilendirmeleri konusunda uyar�lmal�d�rlar.
B�brek fonksiyonu normal olan hastalar ile b�brek yetmezli�i olan hastalar aras�nda advers etkilerin s�kl��� a��s�ndan klinik bir farkl�l�k olmamas�na ra�men, azasitidin ve/veya metabolitleri esas olarak b�brekten at�ld��� i�in b�brek yetmezli�i olan hastalar yak�ndan izlenmelidir (bkz. B�l�m 4.2)
Laboratuvar Testleri:
Tedaviye ba�lamadan ve her tedavi siklusundan �nce karaci�er fonksiyon testleri, serum kreatinin ve serum bikarbonat d�zeyleri belirlenmelidir.
Tedaviye ba�lamadan ve en az her tedavi siklusundan �nce, cevap ve toksisiteyi izlemek
gerekli oldu�u i�in tam kan say�mlar� yap�lmal�d�r (bkz. B�l�m 4.8). Kalp ve akci�er hastal���
Ciddi konjestif kalp yetmezli�i, klinik olarak stabil olmayan kalp hastal��� veya akci�er hastal��� olan hastalar Azasitidin endikasyon �al��malar�na (AZA PH GL 2003 CL 001 ve AZA- AML-001) al�nmam��t�r ve bu y�zden VIDAZA'n�n bu hastalarda g�venlili�i ve etkilili�i saptanamam��t�r. Bilinen bir kalp veya akci�er hastal��� ge�mi�i olan hastalarda yap�lan bir klinik �al��madan al�nan yeni veriler, VIDAZA ile kardiyak olaylar�n insidans�nda �nemli bir art�� oldu�unu g�stermi�tir (bkz. B�l�m 4.8). Bu nedenle, bu hasta grubunda VIDAZA kullan�rken dikkatli olunmas� �nerilir. VIDAZA ile tedavi �ncesinde ve tedavi s�ras�nda kardiyopulmoner de�erlendirme yap�lmas� d���n�lmelidir.
Nekrotizan fasiit
VIDAZA ile tedavi edilen hastalarda, �l�mc�l vakalar da dahil olmak �zere nekrotizan fasiit rapor edilmi�tir. Nekrotizan fasiit geli�en hastalarda, VIDAZA tedavisi hemen durdurulmal� ve acilen uygun bir tedaviye ba�lanmal�d�r.
T�m�r lizis sendromu:
Tedavi �ncesinde y�ksek t�m�r y�k� olan hastalar t�m�r lizis sendromu a��s�ndan risk alt�ndad�r. Bu hastalar yak�n takip edilmeli ve uygun �nlemler al�nmal�d�r.
Diferansiyasyon sendromu
Enjekte edilebilir azasitidin alan hastalarda diferansiyasyon sendromu (retinoik asit sendromu olarak da bilinir) vakalar� bildirilmi�tir. Diferansiyasyon sendromu �l�mc�l olabilir ve semptomlar ve klinik bulgular solunum s�k�nt�s�, pulmoner infiltratlar, ate�, d�k�nt�, pulmoner �dem, periferik �dem, h�zl� kilo al�m�, plevral ef�zyonlar, perikardiyal ef�zyonlar, hipotansiyon ve b�brek yetmezli�ini i�erir (bkz. B�l�m 4.8). Y�ksek doz IV kortikosteroidlerle tedavi ve hemodinamik izleme, diferansiyasyon sendromunu d���nd�ren semptom veya bulgular�n ilk ba�lang�c�nda d���n�lmelidir. Semptomlar d�zelene kadar enjekte edilebilir azasitidinin ge�ici olarak kesilmesi d���n�lmeli ve devam edilirse dikkatli olunmas� �nerilmektedir.
4.5. Di�er t�bbi �r�nler ile etkile�imler ve di�er etkile�im �ekilleri
�n vitro verilere g�re sitokrom P450 izoenzimleri (CYP'ler), UDP-glukuronoziltransferazlar (UGT'ler), s�lfotransferazlar (SULT'ler) ve glutatyon transferazlar�n (GST'ler) azasitidin metabolizmas�nda yer almad��� g�r�lmektedir; bu nedenle bu metabolik enzimler ile ili�kili in vivo etkile�im olas�l���n�n olmad��� d���n�lmektedir.
Azasitidinin sitokrom P450 enzimleri �zerinde klinik olarak �nemli inhibit�r veya ind�kleyici etkisi olas� de�ildir (bkz B�l�m 5.2).
Azasitidin ile klinik ila� etkile�me �al��malar� yap�lmam��t�r.
�zel pop�lasyonlara iliskin ek bilgiler
Hi�bir etkile�im �al��mas� yap�lmam��t�r.
Pediyatrik pop�lasyon
Hi�bir etkile�im �al��mas� yap�lmam��t�r.
4.6. Gebelik ve laktasyon
Gebelik kategorisi: D
�ocuk do�urma potansiyeli bulunan kad�nlar/ Do�um kontrol� (Kontrasepsiyon)
�ocuk do�urma potansiyeli olan kad�nlar tedavi s�ras�nda ve tedaviden sonra en az 6 ay boyunca etkili kontrasepsiyon y�ntemi kullanmal�d�rlar. Erkeklere tedavi s�ras�nda �ocuk sahibi olmamalar�, tedavi s�ras�nda ve tedaviden sonra en az 3 ay boyunca etkili do�um kontrol y�ntemleri kullanmalar� tavsiye edilmelidir.
Gebelik d�nemi
Azasitidinin, gebe kad�nlarda kullan�m�na ili�kin yeterli veri yoktur. Fareler �zerinde yap�lan �al��malar �reme toksisitesi oldu�unu g�stermi�tir (bkz. B�l�m 5.3). �nsanlar i�in potensiyel riski bilinmemektedir. Azasitidin, hayvan �al��malar�ndan elde edilen sonu�lara ve mekanizmas�na dayanarak gebelik s�ras�nda, �zellikle ilk trimesterde, kesinlikle gerekli olmad�k�a kullan�lmamal�d�r. Tedavinin anne i�in avantajlar� fetus i�in olas� risklerine kar�� her vaka i�in tart���larak karar verilmelidir.
Laktasyon d�nemi
Azasitidin / metabolitlerinin anne s�t�ne ge�ip ge�medi�i bilinmemektedir. Emzirilen bebekte ciddi advers reaksiyon potansiyeli nedeniyle, azasitidin tedavisi s�ras�nda emzirme kontrendikedir.
�reme yetene�i /Fertilite
�nsanlarda azasitidinin fertilite �zerindeki etkisine dair herhangi bir veri yoktur. Hayvanlarda azasitidin kullan�m�n�n erkek fertilitesi �zerinde advers reaksiyonlar� g�r�lm��t�r (bkz. B�l�m 5.3).
Tedaviye ba�lamadan �nce erkek hastalara spermlerini saklamak �zere dan��man aramalar� tavsiye edilmelidir.
4.7. Ara� ve makine kullan�m� �zerindeki etkiler
Azasitidinin ara� veya makina kullan�m�na hafif ve orta derecede etkisi vard�r. Azasitidin kullan�m� ile yorgunluk rapor edilmi�tir. Bu nedenle, ara� veya makine kullan�rken dikkatli olunmas� �nerilmelidir.
4.8. �stenmeyen etkiler
G�venlilik profili �zeti
MDS, KMML ve % 20-30 kemik ili�i blastl� AML'si olan yeti�kinlerde:
Hastalar�n %97'sinde VIDAZA uygulamas� ile ili�kili advers reaksiyonlar olu�mu�tur.
Azasitidin endikasyon �al��mas�nda (AZA PH GL 2003 CL 001) en s�k g�r�len ciddi advers reaksiyonlar febril n�tropeni (%8) ve anemi (%2,3) olup bu �al��may� destekleyen �al��malarda da (CALGB 9221 ve CALGB 8921) benzer ciddi advers reaksiyonlar raporlanm��t�r. Daha az s�kl�kta bildirilen di�er ciddi advers reaksiyonlar n�tropenik sepsis (%0,8) ve bazen �l�mc�l sonu�lar� olabilen pn�moni (%2,5) gibi enfeksiyonlar�, trombositopeni (%3,5), a��r� duyarl�l�k reaksiyonlar� (% 0,25) ve kanama olaylar�n� [�rne�in serebral kanama (%0,5), gastrointestinal kanama (%0,8) ve intrakraniyal kanama (%0,5)] i�ermektedir.
Azasitidin tedavisi ile �ok yayg�n g�r�len advers reaksiyonlar trombositopeni, n�tropeni ve l�kopeniyi (genellikle Derece 3-4) i�eren hematolojik reaksiyonlar (%71,4), bulant�, kusmay� (genellikle Derece 1-2) i�eren gastrointestinal olaylar (%60,6) veya enjeksiyon b�lgesi reaksiyonlar�d�r (%77,1; genellikle Derece 1-2).
% 30'dan fazla kemik ili�i blast� olan 65 ya� ve �st� hastalarda:
Azasitidin tedavi kolunda AZA-AML-001 �al��mas� ile belirlenmi� olan �ok yayg�n ciddi advers reaksiyonlar (≥ %10) aras�nda febril n�tropeni (%25), pn�moni (%20,3) ve pireksi (%10,6) bulunmaktad�r. Ayr�ca daha az s�kl�kla raporlanm�� olan ciddi advers reaksiyonlar aras�nda sepsis (%5,1), anemi (%4,2), n�tropenik sepsis (%3), idrar yolu enfeksiyonu (%3),
trombositopeni (%2,5), n�tropeni (%2,1), sel�lit (%2,1), ba� d�nmesi (% 2,1) ve dispne (%2,1) bulunmaktad�r.
Azasitidin tedavisi ile en s�k raporlanan advers reaksiyonlar (�al��madaki hastalar�n %30'unda g�r�len), kab�zl�k (%41,9), mide bulant�s� (%39,8) ve ishali de i�eren sindirim sistemi olaylar� (%36,9; genellikle Derece 1-2), pireksiyi de i�eren genel bozukluklar ve uygulama b�lgesine ili�kin durumlar (%37,7; genellikle Derece 1-2) ve febril n�tropeni (%32,2) ve n�tropeniyi (%30,1; genellikle Derece 3-4) de i�eren hematolojik olaylard�r
Advers reaksiyonlar�n tablola�t�r�lm�� listesi
A�a��daki tablo azasitidin tedavisi ile ili�kili olabilecek advers reaksiyonlar� i�ermektedir. S�kl�klar, MDS ve AML �zerine yap�lm�� temel klinik �al��malara ve pazarlama sonras� g�zlemlere dayanmaktad�r.
S�kl�klar �u �ekilde tan�mlanm��t�r: �ok yayg�n (≥1/10), yayg�n (≥1/100- <1/10), yayg�n olmayan (≥1/1000- <1/100), seyrek (≥1/10.000- <1/1000), �ok seyrek (<1/10.000), bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor). Her bir s�kl�k grubu i�inde advers reaksiyonlar azalan ciddiyet s�ras�na g�re verilmi�tir. Advers reaksiyonlar, temel klinik �al��malar�n herhangi birinde g�zlemlenen en y�ksek s�kl��a g�re a�a��daki tabloda sunulmaktad�r.
Tablo 1: Azasitidin ile tedavi edilen MDS veya AML hastalar�nda bildirilen advers reaksiyonlar (klinik �al��malar ve pazarlama-sonras� deneyim)
Sistem Organ S�n�f� | �ok yayg�n | Yayg�n | Yayg�n olmayan | Seyrek | Bilinmiyor |
Enfeksiyonlar ve enfestasyonlar | Pn�moni* (bakteriyel, viral ve fungal dahil) Nazofarenjit | Sepsis* (Bakteriyel, viral ve fungal dahil) N�tropenik sepsis* |
|
| Nekrotizan fasiit* |
|
| Solunum yollar� enfeksiyonu (�st solunum yollar� ve bron�it dahil) |
| ||
|
| �drar yolu enfeksiyonlar� |
| ||
|
| Sel�lit |
|
Sistem Organ S�n�f� | �ok yayg�n | Yayg�n | Yayg�n olmayan | Seyrek | Bilinmiyor |
Enfeksiyonlar ve enfestasyonlar (Devam�) |
| Divertik�l iltihab� Oral fungal enfeksiyon |
|
|
|
| Sin�zit | ||||
| Farenjit Rinit | ||||
| Herpes simplex | ||||
| Deri enfeksiyonu | ||||
�yi huylu ve |
|
|
|
| Diferansiyasyon |
k�t� huylu | sendromu | ||||
neoplazmalar |
| ||||
(kist ve polipler |
| ||||
de dahil olmak |
| ||||
�zere) |
| ||||
Kan ve lenf sistem hastal�klar� | Febril n�tropeni* N�tropeni L�kopeni | Pansitopeni* Kemik ili�i yetmezli�i |
|
|
|
| Trombositopeni |
| |||
| Anemi |
| |||
Ba����kl�k sistemi hastal�klar� |
|
| A��r� duyarl�l�k reaksiyonlar� |
|
|
Metabolizma ve beslenme hastal�klar� | Anoreksi ��tah kayb� Hipokalemi | Dehidratasyon |
| T�m�r lizis sendromu |
|
Psikiyatrik hastal�klar | Uykusuzluk | Konf�zyonel durum Anksiyete |
|
|
|
Sinir sistemi | Ba� d�nmesi | �ntrakraniyal |
|
|
|
hastal�klar� | Ba� a�r�s� | kanama* | |||
|
| Bay�lma | |||
|
| Uyku hali | |||
|
| Letarji |
Sistem Organ S�n�f� | �ok yayg�n | Yayg�n | Yayg�n olmayan | Seyrek | Bilinmiyor |
G�z hastal�klar� |
| G�z kanamas� Konjunktival kanama |
|
|
|
Kardiyak hastal�klar |
| Perikardiyal ef�zyon | Perikardit |
|
|
Vask�ler hastal�klar |
| Hipotansiyon* Hipertansiyon |
|
|
|
| Ortostatik | ||||
| hipotansiyon | ||||
| Hematom | ||||
Solunum, g���s hastal�klar� ve mediyastinal hastal�klar | Dispne Burun kanamas� | Plevral ef�zyon Efor dispnesi Faringolaringeal a�r� |
| �nterstisiyal akci�er hastal��� |
|
Gastrointestin al hastal�klar | �shal, Kusma Kab�zl�k Bulant� Kar�n a�r�s� (�st kar�n a�r�s� ve kar�n rahats�zl��� dahil) | Gastrointestinal kanama* (A��z kanamas� dahil) Hemoroidal kanama Stomatit Di� eti kanamas� |
|
|
|
|
| Dispepsi | |||
Hepato- bilier hastal�klar � |
|
| Karaci�er yetmezli- �i* �lerleyen hepatik koma |
|
|
Deri ve deri alt� doku hastal�klar� | Pete�i V�cudun her yerinde olabilecek ka��nt� D�k�nt� Ekimoz | Purpura Alopesi �rtiker Eritem Mak�ler d�k�nt� | Akut febril n�trofilik dermatoz Piyoderma gangreno- zum |
|
|
Kas-iskelet bozukluklar� ve ba� doku ve kemik hastal�klar� | Artralji Kas-iskelet a�r�s� (s�rt, kemik ve ekstremitede a�r�y� i�eren) | Kas spazmlar� Miyalji |
|
|
|
Sistem Organ S�n�f� | �ok yayg�n | Yayg�n | Yayg�n olmayan | Seyrek | Bilinmiyor |
B�brek ve idrar yolu hastal�klar� |
| B�brek yetmezli�i* Hemat�ri Serum kreatinin d�zeyinde art�� | Renal t�b�ler asidoz |
|
|
Genel bozukluklar ve uygulama b�lgesine ili�kin hastal�klar | Pireksi* Yorgunluk Asteni G���s a�r�s� Enjeksiyon b�lgesinde eritem
Enjeksiyon b�lgesinde a�r�
Enjeksiyon b�lgesinde reaksiyon (spesifik olmayan) | Enjeksiyon b�lgesinde: morarma, hematom, sertle�me, d�k�nt�, ka��nt�, enflamasyon, renk bozulmas�, nod�l ve kanama
K�rg�nl�k Titreme Katater b�lge kanamas� |
| Enjeksiyon b�lgesinde nekroz |
|
Ara�t�rmalar | Kilo kayb� |
|
|
|
|
* = Nadir olarak �l�mc�l vakalar rapor edilmi�tir. a = bkz. B�l�m 4.4
Se�ilen Advers reaksiyonlar�n s�ralanmas�
Hematolojik advers reaksiyonlar
Azasitidin tedavisi ile ili�kili olarak �ok yayg�n rapor edilen (≥%10) hematolojik advers reaksiyonlar, genellikle 3. veya 4. dereceden anemi, trombositopeni, n�tropeni, febril n�tropeni ve l�kopenidir.
Bu olaylar�n olma riski daha �ok ilk 2 siklus s�ras�ndad�r, daha sonra hematolojik fonksiyonun normale d�nd��� hastalarda daha az s�kl�kta olu�ur. �o�u hematolojik advers reaksiyonlar, tam kan say�mlar�n�n rutin olarak izlenmesi ve bir sonraki siklusta azasitidin uygulamas�n�n geciktirilmesi, n�tropeni i�in profilaktik antibiyotikler ve/veya b�y�me fakt�r� deste�i (�rne�in G-CSF) ve anemi veya trombositopeni i�in transf�zyonlar ile gerekti�i gibi tedavi edilmektedir.
Enfeksiyonlar
Miyelosupresyon n�tropeniye ve enfeksiyon riskinin artmas�na neden olabilir. Azasitidin alan hastalarda n�tropenik sepsisi de i�eren sepsis ve pn�moni gibi ve baz�lar� �l�mc�l sonu�lara neden olan ciddi advers reaksiyonlar rapor edilmi�tir. Enfeksiyonlar, n�tropeni i�in anti-enfektif ajanlar ve b�y�me fakt�r deste�i (�rne�in G-CSF) kullan�m� ile kontrol alt�na al�nabilir.
Kanama
Azasitidin alan hastalarda kanama g�r�lebilir. Gastrointestinal kanama ve intrakraniyal kanama gibi ciddi advers reaksiyonlar rapor edilmi�tir. �zellikle daha �nceden trombositopenisi olan veya tedaviye ba�l� trombositopenisi geli�en hastalar, kanama belirtileri ve semptomlar� i�in izlenmelidir.
A��r� duyarl�l�k
Azasitidin alan hastalarda ciddi a��r� duyarl�l�k reaksiyonlar� rapor edilmi�tir. Anafilaktik benzeri reaksiyon durumunda azasitidin tedavisi derhal kesilmelidir ve uygun semptomatik tedavi ba�lat�lmal�d�r.
Deri ve deri alt� doku hastal�klar�
Deri ve deri alt� advers reaksiyonlar�n�n �o�unlu�u enjeksiyon b�lgesi ile ilgilidir. Bu advers reaksiyonlar�n hi�biri azasitidinin kesilmesine veya ana �al��malarda azasitidin dozunun azalt�lmas�na neden olmam��t�r. Advers reaksiyonlar�n�n �o�unlu�u tedavinin ilk 2 siklusu s�ras�nda olmu�tur ve sonraki sikluslar ile azalmaya y�nelmi�tir. Enjeksiyon b�lgesinde d�k�nt�/enflamasyon/pruritus, d�k�nt�, eritem ve deri lezyonu gibi subkutan advers reaksiyonlar, antihistaminikler, kortikosteroidler ve non-steroidal anti-enflamatuarlar (NSAIIler) gibi ila�lar�n birlikte kullan�m�n� gerektirebilir. Bu kutan�z reaksiyonlar, bazen enjeksiyon b�lgesinde olu�an yumu�ak doku enfeksiyonlar�ndan ay�rt edilmelidirler. Pazarlama sonras�ndaki g�zlemlerde; azasitidin ile birlikte nadir vakalarda, �l�me yol a�an sel�lit ve nekrotizan fasiit gibi yumu�ak doku enfeksiyonlar� rapor edilmi�tir. Enfeksiy�z advers reaksiyonlar�n klinik y�netimi i�in 4.8 Enfeksiyonlar b�l�m�ne bak�n�z.
Gastrointestinal advers reaksiyonlar
Azasitidin tedavisi ile �ok yayg�n rapor edilen advers reaksiyonlar kab�zl�k, ishal, bulant� ve kusmad�r. Bu advers reaksiyonlar, bulant� ve kusma i�in anti-emetikler, ishal i�in anti- diyaretikler ve kab�zl�k i�in laksatif ve/veya fe�es yumu�at�c�lar� ile semptomatik olarak tedavi edilmelidirler.
Renal advers reaksiyonlar
Azasitidin ile tedavi edilen hastalarda, serum kreatinin de�erlerinde art�� ve hemat�riden renal t�b�ler asidoz, renal yetmezlik ve �l�me kadar giden derecelerde b�brek bozukluklar� rapor edilmi�tir (bkz. B�l�m 4.4).
Hepatik advers reaksiyonlar
Azasitidin tedavisi s�ras�nda, metastatik hastal��a ba�l� olarak t�m�r y�k� �ok olan hastalarda hepatik yetmezlik, ilerleyen hepatik koma ve �l�m g�zlenmi�tir (bkz. B�l�m 4.4).
Kardiyak olaylar
Kardiyovask�ler veya pulmoner hastal�k ge�mi�i oldu�u bilinen hastalar�n dahil edildi�i bir klinik �al��madan al�nan veriler, VIDAZA ile tedavi edilen yeni AML te�hisi konmu� hastalarda kardiyak olaylarda bir art�� oldu�unu g�stermi�tir (bkz. B�l�m 4.4).
Ya�l� hastalar
85 ya� ve �st� hastalarda azasitidinin g�venlili�i ile ilgili s�n�rl� bilgi bulunmaktad�r (AZA- AML-001 �al��mas�nda tedavi edilen 85 ya� ve �st� hastalarda 14 hasta [%5,9] bulunmaktad�r.)
Pediyatrik pop�lasyon
AZA-JMML-001 �al��mas�nda, 28 pediyatrik hasta (1 aydan 18 ya��na kadar) MDS (n = 10) veya juvenil miyelomonositik l�semi (JMML) (n = 18) i�in VIDAZA ile tedavi edilmi�tir (bkz. B�l�m 5.1).
28 hastan�n t�m� en az 1 advers olay ya�ad� ve 17'si (%60,7) en az 1 tedaviyle ili�kili olay ya�ad�. Genel pediyatrik pop�lasyonda en s�k bildirilen advers olaylar ate�, anemi, trombositopeni ve febril n�tropeni dahil hematolojik olaylar ile kab�zl�k ve kusma dahil gastrointestinal olaylard�r.
Klinik �al��madaki �� (3) hasta, ilac�n kesilmesine neden olan tedavi ili�kili olay ya�ad� (ate�, hastal���n ilerlemesi ve kar�n a�r�s�).
AZA-AML-004 �al��mas�nda, molek�ler relapsl� 7 pediyatrik hasta (2-12 ya� aras�), ilk tam remisyondan [CR1] VIDAZA ile tedavi edilmi�tir (bkz. B�l�m 5.1).
7 hastan�n t�m�, tedaviye ba�l� en az 1 advers olay ya�am��t�r. En s�k bildirilen yan etkiler n�tropeni, bulant�, l�kopeni, trombositopeni, diyare ve alanin aminotransferaz (ALT) art���d�r. �ki hasta, dozun kesilmesine yol a�an ate�li n�tropeni, n�tropeni) tedaviyle ili�kili bir olay ya�am��t�r.
Klinik �al��ma s�ras�nda VIDAZA ile tedavi edilen s�n�rl� say�da pediyatrik hastada yeni g�venlilik sinyali tespit edilmemi�tir. Genel g�venlik profili, yeti�kin pop�lasyonun g�venlilik profiliyle tutarl�d�r.
��pheli advers reaksiyonlar�n raporlanmas�
Ruhsatland�rma sonras� ��pheli ila� advers reaksiyonlar�n�n raporlanmas� b�y�k �nem ta��maktad�r. Raporlama yap�lmas�, ilac�n yarar/risk dengesinin s�rekli olarak izlenmesine olanak sa�lar. Sa�l�k mesle�i mensuplar�n�n herhangi bir ��pheli advers reaksiyonu T�rkiye Farmakovijilans Merkezi (T�FAM)'ne bildirmeleri gerekmektedir (www.titck.gov.tr; e- posta: tufam@titck.gov.tr; tel: 0 800 314 00 08; faks: 0 312 218 35 99).
4.9. Doz a��m� ve tedavisi
Klinik �al��malar s�ras�nda azasitidin ile doz a��m� bir vakada rapor edilmi�tir. Hasta, �nerilen ba�lang�� dozunun neredeyse 4 kat� olan, yakla��k 290 mg/m2 tek bir i.v. dozu ald�ktan sonra, hastada ishal, bulant� ve kusma g�r�lm��t�r.
Doz a��m� durumunda, hasta uygun kan say�mlar� yap�larak izlenmeli ve gerekli oldu�u �ekilde destekleyici tedavi almal�d�r. Azasitidinin doz a��m� i�in bilinen spesifik bir antidot yoktur.
��ne ve ��r�nga, azasitidin flakonundan ��kar�ld�ktan sonra azasitidin flakonu kuvvetle �alkalanarak bulan�k, homojen bir s�spansiyon elde edilmelidir. Bu noktada s�spansiyonun her mL'sinde 25 mg azasitidin (100 mg/4 mL) bulunur. Olu�an ila� homojen, bulan�k bir s�spansiyondur, herhangi bir topak i�ermemelidir. E�er b�y�k partik�l veya topak mevcutsa �r�n at�lmal�d�r. Etkin maddeyi uzakla�t�rabilece�i i�in s�spansiyonu filtre etmeyiniz. Baz� adapt�rlerde, ��r�ngalarda ve doz sistemlerinde filtrelerin bulundu�u dikkate al�nmal�d�r. Bu nedenle, bu tip sistemler ila� haz�rland�ktan sonra uygulama i�in kullan�lmamal�d�r.
5. FARMAKOLOJ�K �ZELL�KLER
5.1. Farmakodinamik �zellikler
Farmakoterap�tik grup: Antineoplastik maddeler. Pirimidin analoglar�. ATC kodu: L01BC07
Etki mekanizmas�:
Azasitidinin antineoplastik etkilerini, kemik ili�indeki anormal hematopoietik h�creler �zerinde sitotoksisite ve DNA'n�n hipometilasyonu da dahil olmak �zere �oklu mekanizmalar ile g�sterdi�ine inan�lmaktad�r. Azasitidinin sitotoksik etkileri �u mekanizmalardan kaynaklan�yor olabilir: DNA, RNA ve protein sentezinin inhibisyonu, RNA ve DNA'yla birle�me ve DNA y�k�m yolaklar�n�n aktivasyonu. Non-proliferatif h�creler azasitidine g�receli olarak diren�lidir. Azasitidinin DNA'ya kat�l�m� DNA metiltransferazlar�n�n inaktivasyonu ve DNA'n�n hipometilasyonu ile sonu�lan�r. Normal h�cre siklusu kontrol�, diferansiyasyonu ve �l�m yolaklar�nda g�rev alan anormal derecede metillenmi� genlerin DNA hipometilasyonu, genlerin yeniden ekspresyonu ve kanser- bask�lay�c� fonksiyonlar�n tamiri ile sonu�lanabilir. DNA hipometilasyonu ile azasitidinin sitotoksik veya di�er aktivitelerinin klinik sonu�lar �zerindeki g�receli �nemleri hen�z bilinmemektedir.
Klinik etkililik ve g�venlilik:
MDS, KMML ve kemik ili�inde % 20-30 blast olan AML tan�l� yeti�kinlerde
VIDAZA'n�n etkilili�i ve g�venlili�i uluslararas�, �ok merkezli, kontroll�, a��k-u�lu, randomize, paralel gruplu, Faz 3 kar��la�t�rmal� ara�t�rmada (AZA PH GL 2003 CL 001) incelenmi�tir. Ara�t�rmaya Uluslararas� Prognostik Skorlama Sistemine (UPSS) g�re intermediate-2 ile y�ksek riskli MDS ve Frans�z Amerikan �ngiliz (FAB) s�n�fland�rma sistemine g�re ise RAEB, RAEB-T (%21-30 blast) ile mKMML olan MDS hastalar� dahil edilmi�, sekonder MDS'si olan hastalar ara�t�rmaya dahil edilmemi�tir. Azasitidin (n = 179) konvansiyonel tedavi rejimleri (n = 179) ile kar��la�t�r�lm��t�r. Konvansiyonel tedavi rejimleri, tek ba��na destek tedavi (n = 105), d���k doz sitarabin ve beraberinde destek tedavi (n = 49) veya standart ind�ksiyon kemoterapi ile destek tedaviden (n = 25) olu�mu�tur. Hastalar randomizasyondan �nce doktorlar� taraf�ndan 3 konvansiyonel tedavi rejiminden bir tanesine se�ilmi�lerdir. Hasta VIDAZA grubuna randomize olmam��sa, bu �nceden se�ilen rejimi alm��t�r. Hastan�n ara�t�rmaya dahil edilmesi i�in gereken kriterlerden bir tanesi de “Eastern Cooperative Oncology Group” (ECOG) performans�n�n 0-2 aras�nda olmas�d�r. Sekonder MDS'si olan hastalar ara�t�rmaya dahil edilmemi�tir. Ara�t�rman�n primer sonlan�m noktas� toplam sa� kal�m s�residir. VIDAZA medyan 9 siklus (1-39 siklus aral���nda) ve ortalama 10,2 siklus olacak �ekilde 7 g�n boyunca g�nl�k 75 mg/m2 subkutan dozda uygulanm�� ve 21 g�n ara verilmi�tir (28 g�nden olu�an tedavi siklusu). Tedavi Ama�l� Pop�lasyonda (ITT) ya� ortalamas� 69'dur (38-88 ya� aras�).
358 hasta (179 azasitidin ve 179 konvansiyonel tedavi rejimleri �zerinde yap�lan ITT analizinde, VIDAZA ile medyan 24,46 ayl�k bir sa� kal�ma kar��, konvansiyonel tedavi rejimi tedavisinde 15,02 ayl�k sa� kal�m oldu�u saptanm��t�r. Aradaki fark 9,4 ayd�r. (p<0,0001). Azasitidin kullanan hastalarda iki y�ll�k sa� kal�m oran� %50,8 iken; konvansiyonel tedavi rejimi hastalar�nda %26,2'dir (p< 0,0001).
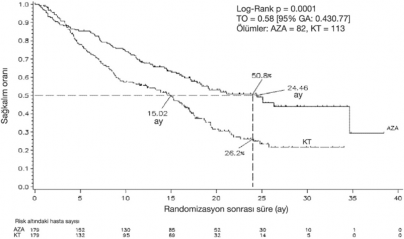
ANAHTAR: AZA= azasitidin; KT= konvansiyonel tedavi; GA= g�venlilik aral���; TO= tehlike oran�
VIDAZA'n�n sa�kal�m faydalar�, kontrol kolunda kullan�lan konvansiyonel tedavi rejimi se�ene�inden (tek ba��na en iyi destek tedavi, d���k doz sitarabin ve beraberinde en iyi destek tedavi veya standart ind�ksiyon kemoterapisi ve beraberinde en iyi destek tedavi) ba��ms�z olarak tutarl�d�r.
UPSS (Uluslararas� Prognostik Skorlama Sistemi) sitogenetik alt grup analiz edildi�inde, t�m gruplarda (iyi, orta, k�t� sitogenetikli, monozomi 7 dahil) medyan genel sa� kal�m a��s�ndan benzer sonu�lar. Ya� alt gruplar� analiz edildi�inde, t�m gruplarda medyan genel sa� kal�mda bir art�� g�zlendi (<65 ya�, ≥65 ya� ve ≥75 ya�).
VIDAZA grubunda �l�m veya AML'ye d�n���m i�in ge�en medyan s�re 13ay iken; bu s�re konvansiyonel rejim tedavisi alan grupta 7,6 ayd�r. VIDAZA 5.4 ayl�k avantaj sa�lam�� olup, p- de�eri 0,0025'dir. Ayr�ca, VIDAZA tedavisi sitopeni ve semptomlar�nda azalma ile birliktelik g�stermi�tir. VIDAZA tedavisi, k�rm�z� kan h�cresi (KKH) ve trombosit transf�zyonlar�na olan ihtiyac�n azalmas�na yol a�maktad�r. Ba�lang��ta KKH transf�zyonuna ba��ml� olan azasitidin grubundaki hastalar�n %45.0'i tedavi s�resi boyunca KKH transf�zyonundan ba��ms�z hale gelirken, kombine CCR gruplar�ndaki ((%33,6 (%95 GA: 22,4, 44,6) istatistiksel olarak anlaml� (p < 0,0001) fark)) hastalar�n %11.4'�nde fark %33.6'd�r. Ba�lang��ta KKH transf�zyonuna ba��ml� olan ve ba��ms�z hale gelen hastalarda, azasitidin grubunda KKH transf�zyon ba��ms�z hale gelme medyan s�resi 13 ayd�r.
Azasitidin grubunda elde edilen toplam yan�t (tam remisyon [TR] + parsiyel remisyon [PR])
%29 iken kombine konvansiyonel tedavi rejimleri grubunda ise %12'dir (p = 0,0001). Ba��ms�z �nceleme Komitesi'nin AZA PH GL 2003 CL1 �al��mas�nda elde etti�i genel yan�t (TR + PR), azasitidin grubunda %7 (12/179) olup bu oran kombine konvansiyonel tedavi gruplar�nda %1 (2/179)'dur (p=0,0113). Ba��ms�z �nceleme Komitesi ve ara�t�rmac� de�erlendirmeleri yan�tlar� aras�ndaki farklar periferik kan say�mlar�n�n iyile�tirilmesini ve en az 56 g�n bu iyile�tirmenin idamesini gerektiren Uluslararas� �al��ma Grubu (IWG) kriterlerinin bir sonucudur. Azasitidin tedavisini takiben TR ve PR elde edilemeyen hastalarda da sa� kal�mda avantajg�zlenmi�tir. Ba��ms�z �nceleme Komitesinin yapt��� de�erlendirmeye g�re azasitidin alan hastalar�n %49'unda hematolojik iyile�me (major veya min�r) tespit edilmi� olup bu oran kombine konvansiyonel tedavi rejimleri ile tedavi edilen hastalarda
%29'dur (p< 0,0001).
Ba�lang��ta bir veya daha fazla sitogenetik anormalli�i olan hastalarda, major sitogenetik yan�t g�r�len hastalar�n oran� azasitidin ve kombine konvansiyonel tedavi rejimi gruplar�nda birbirine benzerdir. Min�r sitogenetik yan�t, kombine konvansiyonel tedavi rejimi grubu ile kar��la�t�r�ld���nda (%10), azasitidin grubunda (%34) istatistiksel olarak anlaml� d�zeyde daha y�ksektir (p = 0,0015).
%30'dan fazla kemik ili�i blast� olan 65 ya� ve �st� akut miyeloid l�semi (AML) hastalar�
AZA-AML-001 klinik ara�t�rmas�nda yer alan tedavi ama�l� hasta pop�lasyonuna ait sonu�lar a�a��da sunulmu�tur (Bkz. 4.1- Terap�tik Endikasyonlar).
V�DAZA'n�n etkililik ve g�venlili�i hematopoietik k�k h�cre transplantasyonuna uygun olmayan, D�nya Sa�l�k �rg�t� s�n�fland�rmas�na g�re 65 ya� ve �st� yeni te�hi� konmu� veya
% 30'dan fazla kemik ili�i blastl� ikincil AML'si olan hastalarda uluslararas�, �ok merkezli, kontroll�, a��k-u�lu, paralel grup Faz 3 �al��mas� yap�lm��t�r. V�DAZA ile birlikte en iyi destek tedavileri (n = 241) konvansiyonel tedavi rejimleri ile kar��la�t�r�lm��t�r. Konvansiyonel tedavi rejimleri, tek ba��na destek tedavileri (n = 45), d���k doz sitarabin ve beraberinde destek tedavileri (n = 158) veya sitarabin ve antrasiklin ile birlikte standart yo�unla�t�r�lm�� kemoterapi ile beraber destek tedaviden (n = 44) olu�maktad�r. Randomizasyondan �nce konvansiyonel tedavi rejimi alan 3 hastadan 1'i doktorlar� taraf�ndan se�ilmi�lerdir. Hastalar e�er V�DAZA grubuna randomize edilmediyse �nceden se�ilmi� tedavi rejimini almaya devam etmi�tir. �al��maya al�nma kriterleri, hastalar�n ECOG performans durumlar�n�n 0 ila
2 aras�nda olmas� ve orta dereceli veya d���k riskli sitogenetik anormalli�i olmas�yd�. �al��man�n birincil sonlan�m noktas� genel sa�kal�m olarak belirlenmi�tir.
VIDAZA alanlar i�in, 21 g�n dinlenme periyodunu takiben 7 g�n boyunca (28 g�nl�k tedavi siklusu) 75 mg/m2 subkutan medyan 6 siklus (1-28 siklus) olacak �ekilde uygulan�rken, sadece en iyi destek tedavisi alanlarda medyan 3 siklus (1-20 siklus), d���k doz sitarabin alanlarda medyan 4 siklus (1-25 siklus) ve standart yo�unla�t�r�lm�� kemoterapi alanlarda medyan 2 siklus (1-3 ind�ksiyon siklusu art� 1 veya 2 konsolidasyon siklusu) olacak �ekilde uygulanm��t�r.
Bireysel ba�lang�� parametreleri a��s�ndan VIDAZA ile konvansiyonel tedavi rejimindeki gruplar kar��la�t�r�labilirdir. Hastalardaki medyan ya� 75'tir (64 ile 91 ya� aral���). %75,2'si beyaz �rktan, %59'u erkek hastalardan olu�maktad�r. D�nya Sa�l�k �rg�t� s�n�fland�rmas�na g�re ba�lang��ta hastalar�n %60,7'si tek ba��na AML, %32,4'� miylodisplaziye ba�l� de�i�iklikler ile AML, %4,1'i terapiye ba�l� miyeloid neoplazma ve %2,9'u tekrar eden genetik anormallikleri ile birlikte AML olarak kategorize edilmi�tir.
488 hastan�n ITT analizinde (241 hasta VIDAZAve 247 hasta konvansiyonel tedavi rejimi ile tedavi edilmi�tir.), VIDAZA tedavisi alan hastalar ile konvansiyonel tedavi rejimi alan hastalar da medyan sa�kal�m oran� s�ras�yla 10,4 ay ve 6,5 ayd�r. Aradaki fark 3,8 ayd�r (p=0,1009). Tedavi etkisinin risk oran� 0,85'tir (%95 GA=0,69; 1,03). Bir y�ll�k sa�kal�m oranlar� VIDAZA alan hastalarda %46,5, konvansiyonel tedavi rejimi alan hastalarda
%34,3't�r.
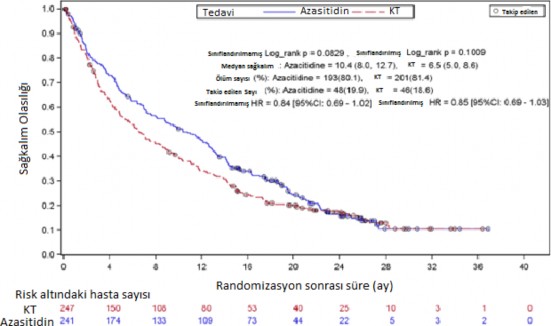
�nceden tan�mlanm�� ba�lang��taki prognostik fakt�rler i�in Cox PH modele uyarlanarak VIDAZA'n�n konvansiyonel tedavi rejimleri kar��la�t�rmas� i�in risk oran� 0,8 (%95 GA=0,66; 0,99; p=0,0355) olarak belirlenmi�tir.
Buna ek olarak, azasitidin ile �nceden se�ilmi� konvansiyonel tedavi rejimi alan hastalar kar��la�t�r�ld���nda �al��ma istatistiksel olarak belirli bir fark g�stermemesine ra�men, VIDAZA kullanan hastalar�n sa�kal�m oran� konvansiyonel tedavi rejimi se�eneklerinden destek tedavisi ve d���k doz sitarabin art� destek tedavisi alan hastalardan daha uzundur. Yo�un kemoterapi ile destek tedavisi alan hastalar ile kar��la�t�r�ld���nda ise sa�kal�m oran� benzerlik g�stermektedir.
VIDAZA'n�n lehine toplam sa�kal�m yarar� y�n�nden, b�t�n �nceden se�ilmi� alt gruplarda ya� [-75 ya� alt� ve 75 ya� ve �st�], cinsiyet, �rk, ECOG performans durumu [0 veya 1 ve 2], temel sitogenetik risk [orta veya d���k], co�rafik b�lge, AML'nin DS� s�n�fland�rmas� (miyelodisplaziye ba�l� de�i�iklikler ile birlikte AML'yi de i�eren), ba�lang��taki l�kosit say�s� [≤ 5 x109/L ve >5 x 109/L], ba�lang��taki kemik ili�i blast� [%50 ve daha az ve >
%50'den �ok], �nceki MDS ge�mi�i] bir e�ilim bulunmaktad�r. Yaln�zca �ok k���k bir grupta toplam sa�kal�m risk oran� istatiksel anlaml�l��a ula�m��t�r. Bu gruplar aras�nda zay�f sitogenetik riski olan hastalar, miyelodisplaziye ba�l� de�i�iklikler olan AML hastalar�, 75 ya� alt� hastalar, kad�n hastalar ve beyaz �rktan hastalar yer almaktad�r.
Hematolojik ve sitogenetik cevaplar ara�t�rmac�lar ve IRC taraf�ndan benzer sonu�lar ile de�erlendirilmi�lerdir. IRC taraf�ndan tam yan�tlar�n oran� (tam remisyon [CR] ve kan say�m� d�zelmesiz tam remisyon [CRi]) VIDAZA grubu i�in % 27,8, ve birle�tirilmi� konvansiyonel tedavi rejimi i�in % 25,1 olarak belirlenmi�tir (p=0,5384). CR ve Cri'ye ula�an hastalarda, remisyon i�in medyan s�re VIDAZA kullanan hastalarda 10,4 ay (% 95 GA =7,5; 15,2) olup , konvansiyonel tedavi rejimi alan hastalarda ise 12,3 ayd�r (% 95 GA =9; 17). VIDAZA ile tedavi edilen ve tam yan�t sa�lanamayan hastalarda konvansiyonel tedavi rejimlerine g�re sa� kal�m avantaj� g�sterilmi�tir.
VIDAZA tedavisi periferik kan de�erlerini iyile�tirmi� ve eritrosit ve trombosit transf�zyonu ihtiyac�n� azaltm��t�r. E�er hasta s�ras�yla 56 g�n (8 hafta) boyunca veya randomizasyon �ncesi bir veya daha fazla eritrosit veya trombosit transf�zyonu alm��sa, ba�lang��ta eritrosit veya trombosit transf�zyonuna ba��ml� kabul edilmi�tir. E�er hasta s�ras�yla tedavi s�resi boyunca ve raporlama periyodunda ard���k gelen herhangi 56 g�n boyunca eritrosit veya trombosit transf�zyonu alm�yorsa, eritrosit veya trobosit transf�zyonuna ba��ml� olmad��� d���n�lmektedir.
Ba�lang��ta eritrosit transf�zyonuna ba��ml� olan VIDAZA gurubundaki hastalardan
%38,5'inin (%95 GA=31,1; 46,2) tedavi periyodu s�resince eritrosit transf�zyonuna ba��ml�l��� kalmam��t�r. Birle�tirilmi� konvasiyonel tedavi rejimi alan hastalarda bu oran
%27,6'd�r (%95 GA=20,9; 35,1). Ba�lang��ta eritrosit transf�zyonuna ba��ml� olan ve tedavi ile transf�zyona ba��ms�z hale gelen hastalar i�in, transf�zyona ba��ms�z hale gelmek i�in ge�en medyan s�re VIDAZA gurubunda 13,9 ay iken konvansiyonel tedavi rejimi alan hastalarda ise bu s�reye ula��lamam��t�r.
�al��ma ba�lang�c�nda trombosit transf�zyonuna ba��ml� olan VIDAZA gurubundaki hastalardan %40,6's�n�n (%95 GA=30,9; 50,8) tedavi periyodu s�resince trombosit transf�zyonuna ba��ml�l��� kalmam��t�r. Birle�tirilmi� konvasiyonel tedavi rejimi alan hastalarda bu oran %29,3't�r (%95 GA=19,7; 40,4). Ba�lang��ta trombosit transf�zyonuna ba��ml� olan ve tedavi ile transf�zyona ba��ms�z hale gelen hastalar i�in, transf�zyona ba��ms�z hale gelmek i�in ge�en medyan s�re V�DAZA gurubunda 10,8 ay iken konvansiyonel tedavi rejimi alan hastalarda ise bu s�re 19,2 ayd�r.
Sa�l��a Ba�l� Ya�am Kalitesi (HRQoL), Avrupa Organizasyonu Kanser Ara�t�rma ve Tedavi �ekirdek Ya�am Kalitesi (EORTC QLQ-C30) anketi kullan�larak belirlenmi�tir. HRQoL verileri test �al��mas�ndaki b�t�n pop�lasyonun alt k�mesi i�in analiz edilebilir. Analizde baz� s�n�rlamalar olmas�na ra�men, elde bulunan veriler VIDAZA tedavisi s�ras�nda hastalar�n ya�am kalitesinde anlaml� bir kay�p ya�amad�klar�n� g�stermektedir.
Pediyatrik pop�lasyon
AZA-JMML-001 �al��mas�, yeni tan� alm�� ileri MDS veya JMML'li pediyatrik hastalarda HSCT'den �nce VIDAZA'n�n farmakokineti�ini, farmakodinami�ini, g�venli�ini ve aktivitesini de�erlendirmek i�in ger�ekle�tirilmi�; Faz 2, uluslararas�, �ok merkezli, a��k etiketli bir �al��mad�r. Klinik �al��man�n birincil amac� VIDAZA'n�n 3. siklus, 28. g�nde yan�t oran� �zerindeki etkisini de�erlendirmektir.
Hastalar (MDS, n = 10; JMML, n = 18, 3 ayl�k – 15 ya� aras�; %71 erkek), minimum 3 siklus ve maksimum 6 siklus boyunca 28 g�nl�k bir siklusun ilk 7 g�n� boyunca, g�nl�k 75 mg/m2 intraven�z VIDAZA dozu ile tedavi edilmi�tir.
MDS koluna hasta al�m�, 10 MDS hastas�ndan sonra etkililik g�zlenmemesi nedeniyle durdurulmu�tur: bu 10 hastada do�rulanm�� yan�t kaydedilmemi�tir.
JMML �al��ma kolunda, 18 hasta (13 PTPN11, 3 NRAS, 1 KRAS somatik mutasyonu ve 1 n�rofibromatozis tip 1 klinik tan�l� [NF 1]) kaydedildi. On alt� hasta 3 siklus, 5 hasta 6 siklus tedaviyi tamamlad�. Toplam 11 JMML hastas�nda, 3. siklusun 28. g�n�nde klinik yan�t al�nm��t�r. Bu 11 hastan�n 9'unda (%50) do�rulanm�� bir klinik yan�t g�zlenmi�tir (3 hastada
cCR - do�rulanm�� tam yan�t ve 6 hastada cPR – do�rulanm�� k�smi yan�t). VIDAZA ile tedavi edilen hasta kohortunda, 7 (%43,8) hastada s�rekli trombosit yan�t� (say�mlar ≥ 100 × 109/L) g�zlenmi� ve HSCT'de 7 (%43,8) hasta transf�zyona ihtiya� duymu�tur. 18 hastadan 17'si HSCT'ye ge�mi�tir.
�al��ma tasar�m� nedeniyle (az hasta say�s� ve kar���kl��a neden olan �e�itli fakt�rler), bu klinik �al��madan HSCT �ncesi VIDAZA'n�n JMML hastalar�nda sa�kal�m� artt�rd��� veya artt�rmad��� sonucu ��kar�lamaz.
AZA-AML-004 �al��mas�, ilk tam remisyondan sonraki molek�ler relaps geli�en AML tan�l� pediyatrik hastalarda ve molek�ler relapstaki AML'li �ocuklarda ve gen� eri�kinlerde anti- kanser tedavisine k�yasla VIDAZA'n�n g�venli�ini, farmakodinami�ini ve etkinli�ini de�erlendirmek i�in bir Faz 2, �ok merkezli, a��k etiketli bir �al��mad�r.
VIDAZA, 7 hastada (ya�lar� 2 ila 12 aras�nda, ortanca 6, 7 y�l ve %71,4'� erkek olan), her 28 g�nl�k siklusun, ilk 7 g�n�nde 100 mg/m2 olacak �ekilde en fazla 3 siklus kullan�lm��t�r.
84. g�nde, 5 hastada minimal rezid�el hastal�k (MRH) de�erlendirmesi yap�ld� ve 4 hastada ya (n=3) molek�ler stabilizasyon ya da (n = 1) molek�ler iyile�me tespit edildi ve bir hastada ise klinik n�ks g�r�ld�. Azasitidin ile tedavi edilen 7 hastan�n alt�s�na (%90 [%95 GA = 0,4; 1]) HKHN uyguland�.
Bu k���k �rneklem b�y�kl��� nedeniyle, VIDAZA'n�n pediyatrik AML'deki etkilili�i belirlenemez.
G�venlilik bilgileri i�in bkz. B�l�m 4.8.
5.2. Farmakokinetik �zellikler
Genel �zelliklerEmilim:
Azasitidin tek 75 mg/m2 subkutan doz uygulamas�ndan sonra, azasitidin 0.5 saatte olu�an (ilk numune alma noktas�) 750 ± 403 ng/mL'lik doruk plazma konsantrasyonlar�yla h�zla absorbe edilmi�tir.
E�ri alt�ndaki alana (EAA) dayanarak subkutan uygulama sonras� azasitidinin I.V. azasitidine (tek 75 mg/m2 doz) g�re biyoyararlan�m� e�ri alt�ndaki alan (EAA) olarak yakla��k %89'dur.
Azasitidinin subkutan uygulamas�n�n e�ri alt�ndaki alan� ve maksimum plazma konsantrasyonu (C) yakla��k 25-100 mg/m2 doz aral��� i�inde orant�l�d�r.
Da��l�m:
IV uygulaman�n ard�ndan ortalama da��l�m hacmi 76 ± 26 L ve sistemik klirensi 147 ± 47 L/saattir.
Biyotransformasyon:
�n vitro verilere g�re sitokrom P450 izoenzimleri (CYPler), UDP-glukuronoziltransferazlar (UGTler), sulfotransferazlar (SULTlar) ve glutatyon transferazlar�n (GSTler) azasitidin metabolizmas�nda yer almad��� g�r�lmektedir.
Azasitidin metabolizmas�, sitidin deaminaz arac�l��� ile olu�an deaminasyon ve spontan olarak geli�en hidroliz ile ger�ekle�mektedir. �nsan karaci�eri S9 fraksiyonlar�nda metabolit olu�umunun NADPH'dan ba��ms�z oldu�u g�zlenmi�tir, bu durum metabolik basamaklar�n sitozolik enzimler taraf�ndan katalizlendi�ine i�aret etmektedir. �nsan hepatosit k�lt�rleri �zerinde yap�lan in vitro ara�t�rmalar 1-100 µM azasitidin konsantrasyonlar�n�n (yani klinik olarak elde edilebilecek konsantrasyonlardan yakla��k 30 kat daha y�ksek konsantrasyonlarda) sitokrom P450 izoenzimleri (CYP) olan “1A2, 2C19 veya 3A4 veya 3A5'i” ind�klemedi�ini g�stermektedir. 100 µM azasitidin ile ink�be edilen bir seri P450 izoenziminde (CYP 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 ve 3A4) inhibisyon olu�turmam��t�r. Bu nedenle klinik olarak elde edilebilir azasitidin plazma konsantrasyonlar�nda enzim inhibisyonu olas�l��� d���n�lmemektedir.
Eliminasyon:
Azasitidin s.c. uygulamadan sonra 41±8 dakikal�k ortalama eliminasyon yar�lanma �mr� tile h�zl� bir �ekilde plazmadan at�l�r. G�nde 1 defa 7 g�n boyunca subkutan 75 mg/m2 azasitidin uygulamas�ndan sonra herhangi bir birikme olu�maz.
Azasitidin ve/veya metabolitleri ba�l�ca idrarla at�l�r.
14C-azasitidinin s.c. ve i.v. uygulamas�n�n ard�ndan, uygulanan radyoaktivitenin <%1'i fe�es ile at�l�rken, % 50-85'i idrar ile at�l�r.
Hastalardaki karakteristik �zellikler
�zel pop�lasyonlar:
Karaci�er yetmezli�inin (bkz B�l�m 4.2), cinsiyetin, ya��n veya �rk�n azasitidinin farmakokineti�i �zerine olan etkileri incelenmemi�tir.
Pediyatrik pop�lasyon
AZA-JMML-001 �al��mas�nda, Farmakokinetik analiz, 1. siklusun 7. g�n�nde 10 MDS ve 18 JMML pediatrik hasta �zerinden ger�ekle�tirilmi�tir. (Bkz. B�l�m 5.1). Ortalama ya� MDS hastalar� i�in 13,3 (ya� aral��� 1,9-15) ve JMML hastalar� i�in de 2,1 (ya� aral��� 0,2-6,9) idi.
75 mg/ m2'lik bir dozun intraven�z uygulanmas�n� takiben VIDAZA, hem MDS hem de JMML pop�lasyonlar�nda 0,083 saat i�inde Cde�erine h�zl� bir �ekilde ula�m��t�r. MDS ve JMML hastalar� i�in C'�n geometrik ortalamas� s�ras�yla 1797,5 ve 1066,3 ng/mL iken AUC'�n geometrik ortalamas� ise 606,9 ve 240,2 ng saat/mL'dir. MDS ve JMML hastalar�nda geometrik ortalama da��l�m hacmi s�ras�yla 103,9 ve 61,1 L'dir. VIDAZA'n�n toplam plazma maruziyetinin MDS hastalar�nda daha y�ksek oldu�u g�r�lm��; bununla birlikte, hem AUC hem de Cde�erleri i�in hastalar aras�nda orta ila y�ksek de�erli de�i�kenlik kaydedilmi�tir.
MDS ve JMML i�in t'nin geometrik ortalamas� s�ras�yla 0,4 ve 0,3 saat ve klerenslerin geometrik ortalamas� ise s�ras�yla 166,4 ve 148,3 L /saat'tir.
AZA-JMML-001 �al��mas�ndan elde edilen farmakokinetik veriler birlikte toplanm�� ve AZA- 2002-BA-002 �al��mas�nda intraven�z yolla 75 mg/m2 dozluk VIDAZA uygulanan MDS'li 6 yeti�kin hastadan al�nan farmakokinetik verilerle kar��la�t�r�lm��t�r. VIDAZA'n�n Cve AUC'nin ortalamas�, intraven�z uygulamadan sonra yeti�kin hastalar ve pediatrik hastalar aras�nda benzerdir (s�ras�yla, 2750 ng/mL'ye kar�� 2841 ng/mL ve 1025 ng∙saat/mL'ye kar��l�k 882,1 ng∙saat/mL).
AZA-AML-004 �al��mas�nda farmakokinetik analiz, yedi hastan�n doz sonras� en az bir
�l��lebilir farmakokinetik konsantrasyon saptanabilmi� alt�s�n�n verileriyle yap�lm��t�r. (bkz. B�l�m 5.1). AML hastalar�n�n medyan ya�� 6,7 ve ya� aral��� ise 2-12 idi.
100 mg/m2'lik �oklu dozun bir �ok kez verilmesi sonras�nda 1.siklusun 7. g�n� Cmax ve AUC0-tau geometrik ortalamalar� s�ras�yla 1557 ng/mL ve 899,6 ng-saat/mL olmu�tur. Hastalar aras� Cmax ve AUC0-tau de�erlerinde y�ksek de�i�kenlik oldu�u g�zlenmi�tir (CV y�zdesi Cmax ve AUC0-tau i�in s�ras�yla %201,6 ve %87,8 olmu�tur). Azasitidin, intraven�z uygulamadan sonra ortalama 0,09 saatlik bir medyan s�rede h�zla Cmax'a ula�m�� ve 0,38 saatlik bir geometrik ortalama yar�lanma �mr� (t) ile azalm��t�r. Klirens ve da��lma hacmi i�in geometrik ortalama s�ras�yla 127,2 L/sa ve 70,2 L'dir.
AML'li �ocuklarda ilk tam remisyon (CR1)'dan sonra molek�ler relapsda g�zlenen farmakokinetik (azasitidin) maruziyet, MDS'li 10 �ocuk ve JMML'li 18 �ocu�un havuzlanm�� verilerinden elde edilen maruziyet ile kar��la�t�r�labilir ve ayr�ca MDS'li yeti�kinlerdeki azasitidin maruziyeti ile kar��la�t�r�labilir seviyededir.
B�brek yetmezli�i
B�brek yetmezli�inin, tek ve �oklu subkutan uygulamalardan sonra azasitidinin farmakokinetik maruziyetinde herhangi bir �nemli etkisi yoktur. Tek 75 mg/m2 subkutan doz uygulamas�ndan sonra, normal b�brek fonksiyonu olan hastalara k�yasla hafif, orta ve ciddi b�brek yetmezli�i olan hastalar�n ortalama maruziyet de�erleri (EAA ve C),s�ras�yla
%11-21, %15-27 ve %41-66 oran�nda artm��t�r. Bununla birlikte, maruziyet, normal b�brek fonksiyonu olan hastalar i�in g�zlenen ayn� genel maruziyet aral���ndad�r. Azasitidin ve/veya metabolitleri esas olarak b�brekten at�ld��� i�in b�brek yetmezli�i olan hastalar�n yak�ndan izlenmesi ko�ulu ile, azasitidin, ba�lang�� doz ayarlamas� olmaks�z�n b�brek yetmezli�i olan hastalara uygulanabilir.
Farmakogenomikler:
Azasitidin metabolizmas� �zerinde bilinen sitidin deaminaz polimorfizmlerinin etkisi incelenmemi�tir.
5.3. Klinik �ncesi g�venlilik verileri
Azasitidin in vitro bakteriyel ve memeli h�cre sistemlerinde hem gen mutasyonlar�n� hem de kromozomal anomalileri ind�kler. Azasitidinin potansiyel karsinojenitesi farelerde ve s��anlarda incelenmi�tir. Azasitidin 52 hafta boyunca haftada 3 defa intraperitonal (i.p.) uyguland���nda, di�i farelerde hematopoetik sistem t�m�rlerini ind�klemi�tir. 50 hafta s�reyle i.p. olarak uygulanan azasitidin ile tedavi edilen farelerde lenforetik�ler sistem, akci�er, s�t bezi ve deri t�m�rlerinin insidans�n�n artt��� g�r�lm��t�r. S��anlarda bir t�m�r olu�turma �al��mas�nda testik�ler t�m�rlerin insidans� artm��t�r.
Farelerde yap�lan ilk embriyotoksisite �al��malar�nda, organogenezis s�ras�nda azasitidinin tek bir i.p. enjeksiyonundan sonra, intrauterin embriyonal �l�m %44 s�kl�kta (artan rezorpsiyon) g�r�lm��t�r.
Azasitidin verilen farelerde, sert dama��n kapanmas� s�ras�nda veya kapanmas�ndan �nce beyinde geli�imsel anormallikler g�r�lm��t�r. S��anlara preimplantasyon s�recinde verildi�inde, azasitidin herhangi bir advers reaksiyon g�stermemi�tir; fakat organogenezis s�ras�nda verildi�inde a��k�a embriyotoksiktir. Organogenezis s�ras�nda s��anlarda meydana gelen fetal anomaliler �unlard�r: MSS anomalileri (eksensefali, ensefalosel), kol-bacak anomalileri (mikromeli, yumru ayak, sindaktili, oligodaktili) ve di�erleri (mikroftalmi, mikrognazi, gastro�izis, �dem ve kaburga anormallikleri).
Azasitidinin, tedavi edilmemi� di�i fare ile �iftle�meden �nce erkek fareye uygulanmas�, fertilite azalmas� ve embriyonik ve postnatal geli�im s�ras�nda yavrunun kayb� ile sonu�lanm��t�r. Erkek s��anlara verilmesi, testis ve epididimislerin a��rl���n�n azalmas�, sperm say�s�n�n azalmas�, gebelik oranlar�n�n azalmas�, �iftle�en di�ilerde embriyolar�n kayb� ve anormal embriyo art��� ile sonu�lanm��t�r (bkz. B�l�m 4.6).
Azasitidin flakonunun plastik kapa�� temizlenmeli ve yeni bir ��r�nga bat�r�lmal�d�r. Flakon ters d�nd�r�lmeli, i�ne ucunun s�v� seviyesinin alt�nda oldu�undan emin olunmal�d�r. ��r�ngan�n pistonu �ekilerek doz i�in gerekli miktarda ila� �ekilmeli ve ��r�ngada hava olmamas�na dikkat edilmelidir. Daha sonra ��r�nga ve i�nesi flakondan ��kar�lmal� ve ��r�ngan�n i�nesi at�lmal�d�r.
6. FARMAS�T�K �ZELL�KLER
6.1. Yard�mc� maddelerin listesi
Mannitol (E421)
6.2. Ge�imsizlikler
Bu �r�n, B�l�m 6.6'da belirtilen t�bbi �r�nler d���ndaki �r�nler ile kar��t�r�lmamal�d�r.
6.3. Raf �mr�
A��lmam�� toz flakonu: 48 ay
Haz�rland�ktan sonra: VIDAZA, buzdolab�nda saklanmayan enjeksiyonluk su ile haz�rland���nda, haz�rlanan t�bbi �r�n 25°C'de 45 dakika ve 2-8°C'de 8 saat s�re ile kimyasal ve fiziksel stabilitesini korur.
Haz�rlanan t�bbi �r�n�n raf �mr� buzdolab�nda (2-8°C) saklanan enjeksiyonluk su ile uzat�labilir. VIDAZA, buzdolab�nda (2-8°C) saklanan enjeksiyonluk21iltere haz�rland���nda, haz�rlanan t�bbi �r�n 2-8°C'de 22 saat s�re ile kimyasal ve fiziksel stabilitesini korur.
Mikrobiyolojik a��dan haz�rlanan �r�n derhal kullan�lmal�d�r. Hemen kullan�lmayacak ise, kullan�m �ncesi saklama s�resi ve ko�ullar� kullan�c�n�n sorumlulu�undad�r ve buzdolab�nda saklanmayan enjeksiyonluk su ile haz�rland���nda 2-8°C'de 8 saatten fazla ve buzdolab�nda (2- 8°C) saklanan enjeksiyonluk su ile haz�rland���nda 2-8°C'de 22 saatten fazla olmamal�d�r.
6.4. Saklamaya y�nelik �zel tedbirler
25°C'nin alt�ndaki oda s�cakl���nda saklay�n�z.
Haz�rlanan t�bbi �r�n�n saklama ko�ullar� i�in B�l�m 6.3'e bak�n�z.
6.5. Ambalaj�n niteli�i ve i�eri�i
Butil kau�uk t�pa ve aluminyum kapak ile kapat�lan, polipropilen plastik d��mesi olan renksiz Tip I 30 mL cam flakon
Ambalaj b�y�kl���: 1 flakon i�inde 100 mg azasitidin.
6.6. Be�eri t�bbi �r�nden arta kalan maddelerin imhas� ve di�er �zel �nlemler
Kullanma talimat�G�venlik i�in �neriler: VIDAZA sitotoksik bir ila�t�r ve di�er potansiyel toksik bile�iklerde oldu�u gibi, azasitidin s�spansiyonlar�n� haz�rlarken ve ta��rken dikkatli olunmal�d�r.
Antikanser ila�lar�n imhas� ve do�ru �ekilde tutulma prosed�rleri uygulanmal�d�r. Haz�rlanan azasitidin s�spansiyonu cilt ile temas ederse, derhal ve iyice su ve sabun ile y�kanmal�d�r. Mukus membranlarla temas eder ise, su ile iyice y�kanmal�d�r.
Haz�rlama prosed�r�:
A�a��daki malzemeler haz�rlanmal�d�r:
Azasitidin flakonu: enjeksiyonluk su flakonu(lar�); steril olmayan cerrahi eldiven;
��r�ngaya yeni bir subkutan i�ne ucu (25 �l�ek �nerilmektedir) tak�l�r. Enjeksiyon b�lgesinde lokal reaksiyon insidans�n� azaltmak i�in i�ne ucu enjeksiyondan �nce temizlenmemelidir.
 Diyabet Hastal���
Diyabet, ins�lin hormonu ile ilgili problemlerden kaynaklanan bir hastal�kt�r.
Diyabet Hastal���
Diyabet, ins�lin hormonu ile ilgili problemlerden kaynaklanan bir hastal�kt�r. |
 G�da Alerjisi
Her y�l milyonlarca insan yiyeceklere alerji g�steriyor.
G�da Alerjisi
Her y�l milyonlarca insan yiyeceklere alerji g�steriyor. |
�LA� E�DE�ERLER�
| E�de�er �la� Ad� | Barkodu | �la� Fiyat� |
|---|---|---|
| AZATU | 8699650271223 | 4,390.95TL |
| AZAVIX | 8699638795260 | |
| AZIDA | 8697943770019 | 5,352.70TL |
| VIDAZA | 8699538774815 | 5,352.70TL |
| VIZADIS | 8699525770684 | 4,622.07TL |
| Di�er E�de�er �la�lar |
 |
Mide Kanseri Mide kanseri genellikle mideyi t�m�yle kaplayan ve mukus �retmekle g�revli h�crelerde ba�lar. Bu kanser tipine adenokarsinom denir. |
 |
Omurilik zedelenmeleri Omurilik zedelenmesini takip eden birka� g�n i�inde, hi�kimse hasarin ne kadar olacagini tahmin edemez. Buradaki sorun, omuriligin herhangi bir zedelenmesinden hemen sonra, bir omurilik sokunun olusmasidir. |
 |
A��z Kanseri A��z kanserinin en yayg�n t�rleri, dudak, dil, di�etidir. Nadiren yanak i�i veya damak b�lgelerini de i�ine al�r. |
�LA� GENEL B�LG�LER�
Er-Kim �la� Sanayi ve Tic. A.�.
| Geri �deme Kodu | A10386 |
| Sat�� Fiyat� | 5352.7 TL [ 22 Apr 2024 ] |
| �nceki Sat�� Fiyat� | 5352.7 TL [ 15 Apr 2024 ] |
| Original / Jenerik | Original �la� |
| Re�ete Durumu | Normal Re�eteli bir ila�d�r. |
| Barkodu | 8699538774815 |
| Etkin Madde | Azacitidine |
| ATC Kodu | L01BC07 |
| Birim Miktar | 100 |
| Birim Cinsi | MG |
| Ambalaj Miktar� | 1 |
| Antineoplastik ve �mm�nomod�lat�r Ajanlar > Antimetabolitler > Azasitidin |
| �thal ( ref. �lke : Italya ) ve Be�eri bir ila�d�r. |