PRADAXA 110 mg 10 sert kapsül Kısa Ürün Bilgisi
{ Dabigatran }
1. BEŞERİ TIBBİ ÜRÜNÜN ADI
PRADAXA® 110 mg sert kapsül2. KALİTATİF VE KANTİTATİF BİLEŞİM
Her kapsül; 110 mg dabigatran eteksilat baz formuna eşdeğer, 126,83 mg beta-alanin, N-[[2-[[[4 (heksiloksi) karbonil] amino] iminometil] fenil] amino] metil]-1-metil-1H-benzimidazol-5-il] karbonil]-N-2-piridinil-, etil ester, metan-sülfonat (metan sülfonik asit tuzu olarak) içermektedir.
Gün batımı sarısı (E 110) 0.003 mg
Yardımcı maddeler için 6.1.’e bakınız.
Yardımcı maddeler için 6.1.’e bakınız.
3. FARMASÖTİK FORMU
Oral uygulama için sert kapsül.
4.1. Terapötik endikasyonlar
PRADAXA® elektif total kalça replasman cerrahisi ya da total diz replasman cerrahisi geçiren erişkin hastalarda venöz tromboembolik olayların önlenmesinde endikedir.
PRADAXA® ayrıca, aşağıdaki risk faktörlerinden bir ya da daha fazlasına sahip, non-valvuler atriyal fibrilasyonlu erişkin hastalarda inme ve sistemik embolizmin önlenmesinde endikedir:
• Önceden geçirilmiş inme, geçici iskemik atak ya da sistemik embolizm
• Sol ventrikül ejeksiyon fraksiyonu < %40
• Semptomatik kalp yetmezliği, > New York Kalp Birliği (NYHA) Sınıf 2
• Yaş > 75
4.2. Pozoloji ve uygulama şekli
Erişkinler:
Pozoloji:
Elektif total kalça ya da diz replasman cerrahisi geçiren erişkin hastalarda venöz tromboembolik olayların (VTE) önlenmesi: Önerilen PRADAXA® dozu, günde bir kez 110 mg’lık 2 kapsül şeklinde alınan 220 mg’dır. Orta derecede böbrek yetmezliği olan hastalarda kanama riski artar. Bu gibi hastalar için önerilen PRADAXA® dozu, günde bir kez 75 mg’lık 2 kapsül şeklinde alınan 150 mg’dır.
Uygulama sıklığı ve süresi:
Diz replasman cerrahisinden sonra VTE’nin önlenmesi: PRADAXA® tedavisi, cerrahi girişimin tamamlanmasından sonraki 1 - 4 saat içinde oral yoldan tek kapsül (110 mg) ile başlatılmalı ve daha sonra toplam 10 gün süreyle günde bir kez 2 kapsül ile sürdürülmelidir. Eğer hemostaz sağlanmamış ise, tedavinin başlatılması ertelenmelidir. Tedavinin cerrahi girişim günü başlamadığı durumlarda, tedavi günde bir kez 2 kapsül ile başlatılmalıdır.
Kalça replasman cerrahisinden sonra VTE’nin önlenmesi: PRADAXA® tedavisi, cerrahi girişimin tamamlanmasından sonraki 1 - 4 saat içinde oral yoldan tek kapsül (110 mg) ile başlatılmalı ve daha sonra toplam 28 - 35 gün süreyle günde bir kez 2 kapsül ile sürdürülmelidir. Eğer hemostaz sağlanmamış ise, tedavi başlangıcı ertelenmelidir. Tedavinin cerrahi girişim günü başlamadığı durumlarda, tedavi günde bir kez 2 kapsül ile başlatılmalıdır.
Bir ya da daha fazla risk faktörüne sahip, non-valvuler atriyal fibrilasyonlu erişkin hastalarda inme ve sistemik embolizmin önlenmesi: Önerilen günlük PRADAXA® dozu, oral yoldan günde iki kez 150 mg sert kapsül şeklinde alınan 300 mg’dır. Tedavi yaşam boyu sürdürülmelidir.
Uygulama şekli:
PRADAXA® sert kapsül aç ya da tok karına, bir miktar su ile birlikte alınmalıdır. Kapsül açılmamalıdır.
Özel popülasyonlara ilişkin ek bilgiler: Böbrek yetmezliği:
4.3. Kontrendikasyonlar
").
Elektif total kalça ya da diz replasman cerrahisi geçiren erişkin hastalarda venöz tromboembolik olayların önlenmesi: Orta derecede böbrek yetmezliği (kreatinin klerensi 30-50 ml/dk) olan hastalarda doz, günde bir kez 75 mg’lık 2 kapsül şeklinde alınan, 150 mg PRADAXA® olarak azaltılmalıdır.
Bir ya da daha fazla risk faktörüne sahip, non-valvuler atriyal fibrilasyonlu erişkin hastalarda inme ve sistemik embolizmin önlenmesi: Doz uyarlaması gerekli değildir; hastalar oral yoldan günde iki kez 150 mg sert kapsül şeklinde alınan 300 mg doz ile tedavi edilmelidir.
Dabigatran diyalizle uzaklaştırılabilir; ancak klinik çalışmalarda bu yaklaşımın kullanılabilirliği konusunda edinilmiş klinik deneyim sınırlıdır.
Karaciğer yetmezliği:
Bir Faz 1 çalışmasında orta derecede karaciğer yetmezliği (Child Pugh B) olan 12 olguda, 12 kontrole kıyasla sistemik dabigatran temasında bir değişiklik görülmemiştir.
Elektif total kalça ya da diz replasman cerrahisi geçiren erişkin hastalarda venöz tromboembolik olayların önlenmesi: Karaciğer enzimlerinde > 2 Normalin Üst Sınırı (NÜS) yükselme görülen hastalar, elektif kalça ya da diz replasman cerrahisinden sonra venöz tromboembolik olayların önlenmesinin incelendiği klinik araştırmalara dahil edilmemişti. Bu hasta alt-grubunda tedavi deneyimi bulunmamaktadır ve bu nedenle böyle bir popülasyonda PRADAXA® kullanımı önerilmemektedir (bkz. Bölüm 4.4 ve 5.2).
Bir ya da daha fazla risk faktörüne sahip, non-valvuler atriyal fibrilasyonlu erişkin hastalarda inme ve sistemik embolizmin önlenmesi: Karaciğer enzimleri > 2 Normal Üst Sınır (NÜS) yüksek olan hastalar, atriyal fibrilasyon ile ilişkili inme ve sistemik embolizmin önlenmesinin araştırıldığı çalışmaya dahil edilmemişti. Bu hasta alt-grubunda tedavi deneyimi bulunmamaktadır ve bu nedenle böyle bir popülasyonda PRADAXA® kullanımı önerilmemektedir (bkz. Bölüm 4.4 ve 5.2).
Pediyatrik popülasyon:
PRADAXA® 18 yaşın altındaki hastalarda incelenmemiştir. Çocuklarda PRADAXA® tedavisi önerilmemektedir.
Geriyatrik popülasyon:
Yaşlı kişilerdeki farmakokinetik çalışmalarda, böbrek fonksiyonlarında yaşa bağlı azalma olan hastalarda, sistemik ilaç temasının arttığı gösterilmiştir (bkz. Böbrek yetmezliğinde doz ve uygulama).
Elektif total kalça ya da diz replasman cerrahisi geçiren erişkin hastalarda venöz tromboembolik olayların önlenmesi: Doz uyarlaması gerekli değildir, hastalar günde bir kez 110 mg’lık 2 kapsül şeklinde alınan, 220 mg PRADAXA® ile tedavi edilmelidir.
Bir ya da daha fazla risk faktörüne sahip, non-valvuler atriyal fibrilasyonlu erişkin hastalarda inme ve sistemik embolizmin önlenmesi: Doz uyarlaması gerekli değildir; hastalar oral yoldan günde iki kez 150 mg sert kapsül şeklinde alınan 300 mg doz ile tedavi edilmelidir.
Diğer:
Vücut ağırlığı:
Doz uyarlaması gerekli değildir.
PRADAXA® ile güçlü P-glikoprotein inhibitörlerinin (amiodaron, kinidin ya da verapamil) eşzamanlı kullanımı:
4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleri
Elektif total kalça ya da diz replasman cerrahisi geçirmiş ve halen PRADAXA® ile tedavi görmekte olan hastalarda verapamil tedavisi başlatılmasından kaçınılmalıdır. PRADAXA® ve verapamil tedavilerinin birlikte başlatılmasından da kaçınılmalıdır.
Bir ya da daha fazla risk faktörüne sahip, non-valvuler atriyal fibrilasyonlu erişkin hastalarda inme ve sistemik embolizmin önlenmesi: Doz uyarlaması gerekli değildir; hastalar oral yoldan günde iki kez 150 mg sert kapsül şeklinde alınan 300 mg doz ile tedavi edilmelidir.
Kanama riski taşıyan hastalar:
Bir ya da daha fazla risk faktörüne sahip, non-valvuler atriyal fibrilasyonlu erişkin hastalarda inme ve sistemik embolizmin önlenmesi: Potansiyel olarak daha yüksek bir majör kanama riski taşıyan hastalar için (örn. yaş >75, CHADS2 skoru >3, orta derecede böbrek yetmezliği (CrCl 3050 ml/dk), ya da güçlü P-gp inhibitörleriyle eşzamanlı tedavi (kinetik bölümündeki Farmakokinetik etkileşimler’e bakınız), ya da önceden gastrointestinal kanama geçirmiş hastalar), günde iki kez 110 mg şeklinde verilen 220 mg’lık azaltılmış bir günlük doz gündeme getirilebilir.
PRADAXA® tedavisinden parenteral antikoagülanlara geçiş:
Elektif total kalça ya da diz replasman cerrahisi geçiren erişkin hastalarda venöz tromboembolik olayların önlenmesi: PRADAXA® tedavisinden parenteral bir antikoagülana geçiş yapılmadan önce, son dozun üzerine 24 saat beklenmelidir.
Bir ya da daha fazla risk faktörüne sahip, non-valvuler atriyal fibrilasyonlu erişkin hastalarda inme ve sistemik embolizmin önlenmesi: PRADAXA® tedavisinden parenteral bir antikoagülana geçiş yapılmadan önce, son dozun üzerine 12 saat beklenmelidir.
Parenteral antikoagülan tedavisinden PRADAXA®’ya geçiş:
PRADAXA® alternatif tedavinin bir sonraki dozunun gelmiş olacağı zamandan 0-2 saat öncesinde, ya da sürekli tedavi durumunda (örn. intravenöz UFH) uygulamanın sonlandırıldığı zaman verilmelidir.
Vit. K antagonistlerinden PRADAXA®’ya geçiş:
Bir ya da daha fazla risk faktörüne sahip, non-valvuler atriyal fibrilasyonlu erişkin hastalarda inme ve sistemik embolizmin önlenmesi: Vit. K antagonisti durdurulmalıdır. INR <2.0 olur olmaz PRADAXA® verilebilir.
Kardiyoversiyon:
Bir ya da daha fazla risk faktörüne sahip, non-valvuler atriyal fibrilasyonlu erişkin hastalarda inme ve sistemik embolizmin önlenmesi: Hastalar kardiyoversiyon uygulanırken PRADAXA® almaya devam edebilirler.
Dozun unutulması:
Elektif total kalça ya da diz replasman cerrahisi geçiren erişkin hastalarda venöz tromboembolik olayların önlenmesi: Bir sonraki gün aynı saatte, kalan günlük PRADAXA® dozlarının alınmasına devam edilmelidir.
Unutulan tekil dozları telafi etmek için çift doz alınmamalıdır.
4.3. Kontrendikasyonlar
• Dabigatran ya da dabigatran eteksilata karşı veya ürünün içindeki yardımcı maddelerden birine karşı bilinen aşırı duyarlılık,
• Şiddetli böbrek yetmezliği (CrCl < 30 ml/dk),
• Aktif, klinik olarak anlamlı kanama,
• Kanama diyatezi olan hastalar, ya da spontan veya farmakolojik hemostaz bozukluğu olan hastalar,
• Klinik olarak anlamlı kanama riski taşıyan organ lezyonları, -son 6 ay içerisinde geçirilmiş hemorajik inme dahil,
4.4. Özel kullanım uyarıları ve önlemleri
• Karaciğer yetmezliği ya da sağkalım üzerinde etkili olması beklenen karaciğer hastalığı,
4.4. Özel kullanım uyarıları ve önlemleri
Karaciğer yetmezliği:
Karaciğer enzimleri > 2 Normal Üst Sınır (NÜS) yüksek olan hastalar, elektif kalça ya da diz replasman cerrahisinden sonra VTE’den korunmanın incelendiği kontrollü klinik araştırmalara ve atriyal fibrilasyon ile ilişkili inme ve sistemik embolizmin önlenmesinin araştırıldığı çalışmaya dahil edilmemişti. Bu hasta alt-grubunda tedavi deneyimi bulunmamaktadır ve bu nedenle böyle bir popülasyonda PRADAXA® kullanımı önerilmemektedir.
Hemorajik risk:
Bütün antikoagülanlar ile olduğu gibi, PRADAXA® kanama riskinin yüksek olduğu durumlarda dikkatle kullanılmalıdır. PRADAXA® tedavisi sırasında herhangi bir bölgede kanama ortaya çıkabilir. Hemoglobin ve/veya hematokrit düzeylerinde ya da kan basıncında açıklanamayan bir düşme, bir kanama bölgesi için araştırma yapılmasına öncülük etmelidir.
aPTT testi yaygın bir şekilde kullanılmaktadır ve PRADAXA® ile sağlanan antikoagülasyon yoğunluğu için yaklaşık bir gösterge sağlar. Kanaması olan hastalarda aPTT testi, duyarlılığı kısıtlı olmakla birlikte, antikoagülan aktivite fazlalığının belirlenmesine yardımcı olarak yarar sağlayabilir. 80 saniyeden daha büyük bir aPTT değeri, daha yüksek bir kanama riskiyle ilişkilidir.
Farmakokinetik çalışmalarda, böbrek işlevlerinde yaşa bağlı azalma dahil olmak üzere, böbrek işlevleri azalmış hastalarda sistemik ilaç temasının arttığı ortaya konulmuştur. PRADAXA® şiddetli böbrek yetmezliği (CrCl <30 mL/dk) olgularında kontrendikedir.
Akut böbrek yetmezliği gelişen hastalarda PRADAXA®’ya son verilmelidir.
4.2. Pozoloji ve uygulama şekli
Aşağıdaki ilaçların PRADAXA® ile eşzamanlı olarak kullanılması incelenmemiştir ve kanama riskini arttırabilir; fraksiyone olmayan heparinler (santral venöz ya da arteriyel kateterde açıklığın sürdürülmesi için gerekli dozlar hariç) ve heparin türevleri, düşük molekül ağırlıklı heparinler (LMWH), fondaparinuks, desirudin, trombolitik ajanlar, GPIIb/IIIa reseptör antagonistleri, tiklopidin, dekstran, sülfinpirazon, rivaroksaban, prasugrel, vitamin K antagonistleri ve P-gp inhibitörleri olan dronedaron, itrakonazol, takrolimus, siklosporin, ritonavir, tipranavir, nelfinavir ve sakinavir.
Kanama riskinin arttığı durumlarda (örn. yakınlarda geçirilmiş biyopsi ya da majör travma, bakteriyel endokardit) yakın gözlem yapılması (kanama ya da anemi işaretlerinin aranması) genellikle gerekli olmaktadır.
Elektif total kalça ya da diz replasman cerrahisi geçiren erişkin hastalarda venöz tromboembolik olayların önlenmesi: Kısa dönemli perioperatif analjezi için verilen NSAEİ’lerin, PRADAXA® ile birlikte uygulandıklarında kanama riskinde artış ile ilişkili olmadıkları gösterilmiştir.
PRADAXA® tedavisi sırasında yarı-ömürleri 12 saatten daha kısa olan NSAEİ’lerin düzenli kullanımına yönelik kısıtlı veri mevcuttur ve burada da ek bir kanama riski izlenimi bulunmamaktadır.
Bir ya da daha fazla risk faktörüne sahip, non-valvuler atriyal fibrilasyonlu erişkin hastalarda inme ve sistemik embolizmin önlenmesi: Birlikte uygulanan oral anti-trombositer (aspirin ve klopidogrel dahil) ve NSAEİ tedavileri kanama riskini arttırır.
P-gp indükleyicileriyle etkileşim:
4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleri
" ve "Hastalardaki karakteristik özellikler").
Cerrahi ve girişimler:
PRADAXA® almakta iken cerrahi ya da invazif prosedürler uygulanan hastalarda kanama riski artar. Bu nedenle, cerrahi girişimlerde PRADAXA®’nın geçici olarak durdurulması gerekli olabilir (aynı zamanda bkz. "Farmakokinetik").
Preoperatif dönem:
İnvazif ya da cerrahi prosedürlerden önce, kanama riskinde artış nedeniyle, PRADAXA® geçici olarak durdurulmalıdır. PRADAXA® eğer mümkünse, invazif ya da cerrahi prosedürlerden en az 24 saat öncesinde sonlandırılmalıdır. Kanama riski daha yüksek olan hastalarda ya da tam bir hemostaza ihtiyaç duyulabilecek majör cerrahilerde, PRADAXA®’ya cerrahiden 2 - 4 gün önce son verilmesi gündeme getirilmelidir. Böbrek yetmezliği olan hastalarda dabigatran klerensi daha uzun zaman alabilir. Herhangi bir prosedür öncesinde bu durum dikkate alınmalıdır (aynı zamanda bkz. "Farmakokinetik").
PRADAXA® şiddetli böbrek disfonksiyonu olan (CrCl <30 mL/dk) hastalarda kontrendikedir, ancak böyle bir tablonun ortaya çıkması halinde, PRADAXA® majör cerrahiden en az 5 gün önce kesilmelidir.
Eğer akut bir girişim gerekiyorsa, PRADAXA® geçici olarak durdurulmalıdır. Cerrahi ya da girişim, eğer mümkünse, son dozdan en az 12 saat sonrasına kadar ertelenmelidir. Eğer cerrahi ertel enemiyorsa, kanama riskinde artış olabilir. Girişimin aciliyeti ile birlikte kanama riski de tartılmalıdır.
Spinal anestezi / Epidural anestezi / Lomber ponksiyon:
Spinal anestezi gibi prosedürler, tam bir hemostatik fonksiyonu gerektirebilir.
Spinal ya da epidural hematom riski, travmatik ya da tekrarlı ponksiyon uygulanan olgularda ve
uzun süreli epidural kateter kullanımı halinde artış gösterebilir. Bir kateterin çıkarılmasının
ardından, ilk PRADAXA® dozu uygulanmadan önce en az 1 saatlik bir aranın geçmesi
gereklidir. Bu hastalar spinal ya da epidural hematomun nörolojik bulgu ve semptomlarına
yönelik sıkı bir gözlem altında tutulmalıdır.
Post-prosedürel dönem:
Tam bir hemostaz sağlandığında, tedavi yeniden başlatılır.
Yardımcı maddeler:
4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleri
Antikoagülanlar ve antitrombositer agregasyon ajanları:
4.4. Özel kullanım uyarıları ve önlemleri
Aşağıdaki ilaçlar incelenmemiştir ve PRADAXA® ile birlikte kullanıldıklarında kanama riskini arttırabilirler: UFH, düşük molekül ağırlıklı heparinler (LMWH) ve heparin türevleri (fondaparinuks, desirudin), trombolitik ajanlar, GPIIb/IIIa reseptör antagonistleri, tiklopidin, prasugrel, dekstran, sülfinpirazon, rivaroksaban ve vitamin K antagonistleri (bkz. Bölüm 4.4).
UFH, santral venöz ya da arteriyel kateterde açıklığın sürdürülmesi için gereken dozlarda kullanılabilir (bkz. Bölüm 4.2 ve 4.4).
Klopidogrel: Sağlıklı genç erkek gönüllüler üzerindeki bir faz I çalışmasında, dabigatran eteksilat ile birlikte klopidogrel kullanımı, kapiller kanama zamanlarında, klopidogrel monoterapisine kıyasla daha fazla bir uzama ile sonuçlanmamıştı. Kombine tedavi ve ilişkili monoterapiler karşılaştırıldığında aynı zamanda, dabigatran EAAT,ss ve Cmaks,ss değerleri, dabigatran etkisinin koagülasyon ölçümleri, ya da klopidogrel etkisinin ölçümü olarak trombosit agregasy onunun inhibisyonu esas olarak değişmeksizin kalmıştı. 300 ya da 600 mg klopidogrel yükleme dozuyla, dabigatran EAAT,ss ve Cmaks,ss değerleri %30 - 40 civarında yükseldi (bkz. Bölüm 4.4).
Asetilsalisilik asit (ASA): Dabigatran eteksilat ile birlikte ASA uygulamasının kanama riski üzerindeki etkisi, atriyal fibrilasyonlu hastalar üzerinde birlikte randomize ASA uygulamasının gerçekleştirildiği bir Faz II çalışmasında incelenmiştir. Lojistik regresyon analizi temelinde, ASA ve günde iki kez 150 mg dabigatran eteksilat uygulaması herhangi bir kanama riskini, 81 mg ve 325 mg ASA ile, %12’den sırasıyla, %18 ve %24’e arttırabilir (bkz. Bölüm 4.4).
Faz III RE-LY çalışmasında toplanan verilerden (bkz. Bölüm 5.1), günde iki kez 110 mg ya da 150 mg dozlarında dabigatran eteksilat ile birlikte ASA ya da klopidogrel uygulamasının majör kanama riskini arttırabileceği gözlenmiştir (bkz. Bölüm 4.4). Birlikte ASA ya da klopidogrel uygulamasındaki daha yüksek oranlarda kanama olayları, aynı zamanda varfarin ile de gözlenmiştir.
NSAEİ: Kısa dönemli perioperatif analjezi için verilen NSAEİ’lerin, dabigatran eteksilat ile birlikte uygulandıklarında, kanama riskinde artış ile ilişkili olmadığı gösterilmiştir. RE-LY çalışmasındaki kronik kullanımda NSAEİ’ler kanama riskini, hem dabigatran hem de varfarin ile yaklaşık %50 arttırmıştır. Bu nedenle, hemoraji riski yüzünden, özellikle eliminasyon yarı-ömürleri 12 saatin üzerinde olan NSAEİ’ler kullanıldığında kanama bulguları için yakın gözlem yapılması önerilmektedir (bkz. Bölüm 4.4).
Düşük molekül ağırlıklı heparinler (LMWH): Enoksaparin gibi LMWH’ların dabigatran eteksilat ile birlikte kullanılması özel olarak araştırılmamıştır. Üç gün süreli, günde bir kez s.c. 40 mg enoksoparin tedavisinden geçiş yapıldığında, son enoksaparin dozundan 24 saat sonra sistemik dabigatran teması, tek başına dabigatran eteksilat (tek doz 220 mg) uygulamasından sonra bulunandan biraz daha düşüktü. Enoksaparin ön tedavisi ile birlikte dabigatran eteksilat uygulamasından sonra, tek başına dabigatran eteksilat uygulamasından sonrasına kıyasla daha yüksek bir anti-FXa/FIIa aktivitesi gözlendi. Bu durumun enoksaparin tedavisinin aktarıcı etkisine bağlı olduğu düşünülmektedir ve klinik anlam taşımadığı kabul edilmektedir. Enoksaparin ön tedavisiyle, dabigatran ile ilişkili diğer antikoagülasyon testlerinde anlamlı değişiklik bulunmamaktaydı.
Dabigatran eteksilat ve dabigatranın metabolik profiline bağlı etkileşimler:
Dabigatran eteksilat ve dabigatran, sitokrom P450 sistemi tarafından metabolize edilmezler ve in vitro insan sitokrom P450 enzimleri üzerinde herhangi bir etkileri yoktur. Bu nedenle, dabigatran eteksilat ya da dabigatran ile, bu sistem ile ilişkili ilaç etkileşimleri beklenmemektedir.
P-gp etkileşimleri: P-glikoprotein inhibitörleri:
Dabigatran eteksilat dışarı akış taşıyıcısı P-gp’nin bir subtratıdır. Güçlü P-gp inhibitörleri (amiodaron, verapamil, kinidin, ketokonazol ve klaritromisin gibi) ile birlikte uygulamanın dabigatran plazma konsantrasyonlarında artış ile sonuçlanması beklenmektedir.
Eğer başka türlü özel bir açıklama bulunmuyorsa, dabigatran güçlü P-gp inhibitörleriyle birlikte uygulandığında, yakın klinik gözetim (kanama ya da anemi bulgularının aranması için) gerekir. Sistemik dabigatran temasında yükselme nedeniyle kanama riskinde artış olan hastaların tanımlanmasında bir koagülasyon testi yardımcı olabilir (bkz. Bölüm 4.2, 4.4 ve 5.1).
Sistemik ketokonazol, itrakonazol, siklosporin ve takrolimus kontrendikedir (bkz. Bölüm 4.3). Diğer güçlü P-gp inhibitörleri (örn. amiodaron, kinidin ya da verapamil) ile birlikte uygulamada dikkatli olunmalıdır (bkz. Bölüm 4.2 ve 4.4).
Ketokonazol: Ketokonazol, total dabigatran EAA0-ro ve Cmaks değerlerini arttırmıştır. Bu artışlar tek doz 400 mg’dan sonra sırasıyla, %138 ve %135, günde bir kez tekrarlı 400 mg ketokonazol dozlarından sonra ise, sırasıyla, %153 ve %149 düzeyindeydi. Doruk konsantrasyon zamanı, terminal yarı-ömür ve ortalama kalış zamanları ketokonazol tarafından etkilenmemişti (bkz. Bölüm 4.4). Sistemik ketokonazol ile birlikte tedavi kontrendikedir (bkz. Bölüm 4.3).
Amiodaron: PRADAXA® oral tek doz 600 mg amiodaron ile birlikte uygulandığında, amiodaron ve aktif metaboliti DEA’nın absorpsiyon hızı ve boyutları esas olarak değişiklik göstermemiştir. Dabigatran EAA ve Cmaks değerleri sırasıyla yaklaşık %60 ve %50 oranında artar. Etkileşimin mekanizması henüz tam olarak aydınlatılmamıştır. Amiodaronun yarı-ömrünün uzun oluşu göz önüne alındığında, ilaç etkileşim potansiyeli, amiodaron kesildikten sonra haftalarca varlığını sürdürebilir (bkz. Bölüm 4.2 ve 4.4).
Kalça ya da diz replasman cerrahisinden sonra VTE’den korunmak için tedavi edilen hastalarda, eğer dabigatran eteksilat ile birlikte amiodaron veriliyorsa, doz günde bir kez 2 kapsül PRADAXA® 75 mg şeklinde uygulanan 150 mg’a azaltılmalıdır (bkz. Bölüm 4.2). Dabigatran eteksilat amiodaron ile kombine edildiğinde, özellikle kanama oluşması durumunda ve aynı zamanda hafif ile orta derecede böbrek yetmezliği olan hastalarda, yakın klinik gözetim önerilmektedir.
Kinidin: Kinidin, total 1000 mg dozuna kadar, iki saatte bir verilen 200 mg’lık dozlar şeklinde uygulandı. Dabigatran eteksilat ardışık 3 gün süreyle, günde iki kez uygulandı ve 3. gün kinidin ile birlikte ya da kinidin olmaksızın verildi. Birlikte verilen kinidin ile, dabigatran EAAT,ss ve Cmaks,ss değerleri, sırasıyla ortalama %53 ve %56 artış gösterdi (bkz. Bölüm 4.2 ve 4.4).
Kalça ya da diz replasman cerrahisinden sonra VTE’den korunmak için tedavi edilen hastalarda, eğer dabigatran eteksilat ile birlikte kinidin veriliyorsa, doz günde bir kez 2 kapsül PRADAXA® 75 mg şeklinde uygulanan 150 mg’a azaltılmalıdır (bkz. Bölüm 4.2). Dabigatran eteksilat kinidin ile kombine edildiğinde, özellikle kanama oluşması durumunda ve aynı zamanda hafif ile orta derecede böbrek yetmezliği olan hastalarda, yakın klinik gözetim önerilmektedir.
Verapamil: Dabigatran eteksilat (150 mg) oral verapamil ile birlikte uygulandığında, dabigatranın Cmaks ve EAA değerleri artış göstermiştir; ancak bu artışın boyutları, verapamilin uygulama zamanına ve formülasyonuna bağlı olarak değişmektedir (bkz. 4.2 ve 4.4).
Dabigatran sistemik temasındaki en büyük artış, dabigatran eteksilatın alınmasından bir saat önce uygulanan çabuk salımlı verapamil formülasyonunun ilk dozunda gözlendi (%180 civarında Cmaks ve %150 civarında EAA artışları). Bu etki uzatılmış salımlı formülasyon uygulamasıyla (%90 civarında Cmaks ve %70 civarında EAA artışları) ya da tekrarlı verapamil doz uygulamasıyla (%60 civarında Cmaks ve %50 civarında EAA artışları) progresif bir şekilde azaldı.
Bu nedenle, dabigatran verapamil ile birlikte uygulandığında, yakın klinik gözetim (kanama ya da anemi bulgularının aranması için) gerekir. Kalça ya da diz replasman cerrahisinden sonra eşzamanlı dabigatran eteksilat ve verapamil alan, böbrek fonksiyonları normal hastalarda PRADAXA® dozu, günde bir kez 75 mg’lık 2 kapsül şeklinde alınan 150 mg’a azaltılmalıdır. Orta derecede böbrek yetmezliği olan ve eşzamanlı dabigatran eteksilat ve verapamil ile tedavi edilen hastalarda PRADAXA® dozunun günde 75 mg’a düşürülmesi gündeme getirilmelidir (bkz. Bölüm 4.2 ve 4.4).
İnme ve sistemik embolizmin önlenmesi için tedavi edilmekte olan ve eşzamanlı dabigatran eteksilat ve verapamil almakta olan non-valvüler atriyal fibrilasyonlu hastalarda PRADAXA® dozu, günde iki kez bir tane 110 mg kapsül şeklinde verilen, 220 mg’a azaltılmalıdır (bkz. Bölüm 4.2).
Dabigatran eteksilat verapamil ile kombine edildiğinde, özellikle kanama oluşması durumunda ve aynı zamanda hafif ile orta derecede böbrek yetmezliği olan hastalarda, yakın klinik gözetim önerilmektedir.
Verapamil dabigatran eteksilattan 2 saat sonra verildiğinde anlamlı bir etkileşim gözlenmemiştir (%10 civarında Cmaks ve %20 civarında EAA artışları). Bu durum, dabigatran emiliminin 2 saat sonra tamamlanmasıyla açıklanmaktadır (bkz. 4.4).
Klaritromisin: Klaritromisin (günde iki kez 500 mg), sağlıklı gönüllülerde dabigatran eteksilat ile birlikte uygulandığında, herhangi bir klinik güvenlilik kaygısı bulunmaksızın, EAA %19 ve Cmaks %15 civarında artmıştır. Ancak, dabigatran almakta olan hastalarda, klaritromisin kombine edildiğinde, klinik açıdan anlamlı bir etkileşim dışlanamaz. Bu nedenle, dabigatran eteksilat klaritromisin ile kombine edildiğinde, özellikle kanama oluşması durumunda ve aynı zamanda hafif ile orta derecede böbrek yetmezliği olan hastalarda, yakın bir izleme yapılmalıdır.
Aşağıdaki güçlü P-gp inhibitörleri klinik olarak incelenmemiştir, ama in vitro sonuçlardan, ketokonazol ile olana benzer bir etki beklenebilir; itrakonazol, takrolimus ve siklosporin. Bunların birlikte kullanılması kontrendikedir.
Posakonazol için ne klinik ne de in vitro test sonuçları bulunmamaktadır ve PRADAXA® ile birlikte kullanılması önerilmemektedir. PRADAXA® ve dronedaronun birlikte kullanılmasına ilişkin klinik veriler yetersizdir ve birlikte kullanılmaları önerilmemektedir (bkz. Bölüm 4.4)
P-glikoprotein indükleyicileri:
Birlikte bir P-gp indükleyicisi (rifampisin, St. John bitkisi (Hypericum perforatum), karbamazepin ya da fenitoin gibi) uygulamasının dabigatran konsantrasyonlarında azalma ile sonuçlanması beklenmektedir ve birlikte uygulamadan kaçınılmalıdır (bkz. Bölüm 4.4 ve 5.2).
Rifampisin: Önceden 7 gün süreyle günde bir kez 600 mg dozunda prob indükleyici rifampisin uygulaması, total dabigatran doruk ve total sistemik temasını, sırasıyla %65.5 ve %67 oranında azaltmıştır. İndükleyici etki daha sonra azaldı ve rifampisin tedavisinin kesilmesinden sonraki 7. günde, referansa yaklaştı. İkinci bir 7 günden sonra, biyoyararlanımda başka artış gözlenmedi.
P-glikoproteini etkileyen diğer ilaçlar:
Ritonavir dahil proteaz inhibitörleri ve bunun diğer proteaz inhibitörleriyle olan kombinasyonları P-gp’yi etkiler (inhibitör ya da indükleyici olarak). Bunlar incelenmemiştir ve PRADAXA® ile birlikte tedavide kullanılmaları önerilmemektedir.
P-glikoprotein substratı:
Digoksin: 24 sağlıklı olguda yürütülen bir çalışmada, PRADAXA® digoksin ile birlikte uygulandığında, digoksinde değişiklik gözlenmemiş ve dabigatran sistemik temasında klinik olarak önemli bir değişiklik izlenmemiştir (bkz. "Farmakokinetik etkileşimler").
Gastrik pH:
Pantoprazol: PRADAXA® pantoprazol ile birlikte uygulandığında, dabigatran plazma konsantrasyon - zaman eğrisi altındaki alanda, yaklaşık %30 civarında bir azalma gözlenmiştir. Klinik araştırmalarda pantoprazol ve diğer proton pompası inhibitörleri (PPİ) dabigatran eteksilat ile birlikte uygulanmış, ve birlikte PPİ tedavisinin PRADAXA®’nın etkinliğini azaltmadığı görülmüştür.
Ranitidin: PRADAXA® ile birlikte ranitidin uygulamasının, dabigatran absorpsiyonu boyutları üzerinde klinik olarak anlamlı bir etkisi bulunmamaktaydı.
Özel popülasyonlara ilişkin ek bilgiler:
Özel bir veri bulunmamaktadır.
Pediyatrik popülasyon:
4.6. Gebelik ve laktasyon
Genel tavsiye:
Gebelik kategorisi C’dir.
Çocuk doğurma potansiyeli bulunan kadınlar/Doğum kontrolü (Kontrasepsiyon): Çocuk doğurma potansiyeli bulunan kadınlar, tedavi süresince tıbben etkili olduğu kabul edilen doğum kontrol yöntemleri kullanmalıdır.
Gebelik dönemi: Gebelikte ilaç temasıyla ilgili klinik veri bulunmamaktadır. İnsanlardaki potansiyel risk bilinmemektedir.
Hayvanlardaki reprodüktif çalışmalarda fertilite ya da yenidoğanın postnatal gelişimi üzerinde herhangi bir advers etki görülmemiştir.
Çocuk doğurma potansiyeline sahip kadınların PRADAXA® tedavisi sırasında gebe kalmaktan kaçınmaları gereklidir ve gebe kadınlar, beklenen yarar risklerinden daha büyük olmadığı sürece, PRADAXA® ile tedavi edilmemelidir.
Laktasyon dönemi: Klinik veri bulunmamaktadır. Bir önlem olarak, süt emzirme durdurulmalıdır.
4.7. Araç ve makine kullanımı üzerindeki etkiler
4.8. İstenmeyen etkiler
PRADAXA®’nın güvenliliği bütünüyle 22.687 hasta üzerinde araştırılmıştır.
Elektif total kalça ya da diz replasman cerrahisinden sonra VTE’den primer korunma araştırmalarında toplam 10,596 hasta, 5 kontrollü çalışma kapsamında en az bir doz çalışma ilacı ile tedavi edilmiştir. Bunların arasında 5,674’ü günde bir kez 150 ya da 220 mg dabigatran eteksilat ile tedavi edilirken, 552’sine günde bir kez 150 mg’dan daha düşük dozlar ve 1168’ine günde bir kez 220 mg’dan daha yüksek dozlar verilmiştir.
Bir ya da daha fazla risk faktörüne sahip, non-valvuler atriyal fibrilasyonlu erişkin hastalarda inme ve sistemik embolizmin önlenmesinin incelendiği RE-LY araştırmasında, toplam 12,091 hasta dabigatran eteksilata randomize edilmiştir. Bunların arasında 6,076’sı günde iki kez 150 mg dabigatran eteksilat ile tedavi edilirken, 6,015’ine günde iki kez 110 mg’lık dozlar verilmiştir.
Toplam olarak, elektif kalça ya da diz cerrahisi için tedavi edilen (42 güne kadar kısa dönemli tedavi) hastaların yaklaşık %9’u ve inme ve sistemik embolizmin önlenmesi için tedavi edilen (3 yıla kadar uzun dönemli tedavi) atriyal fibrilasyonlu hastaların %22’sinde advers reaksiyonlar gelişmiştir.
Kanama:
Kanama, PRADAXA®’nın en önemli yan etkisidir; herhangi bir tipte ve şiddet derecesinde kanama, endikasyona bağlı olarak, elektif kalça ya da diz replasman cerrahisi için kısa süreyle tedavi edilen hastaların yaklaşık %14’ünde ve inme ve sistemik embolizmin önlenmesi için uzun süreyle tedavi edilen atriyal fibrilasyonlu hastaların yıllık %16.5’inde ortaya çıkmıştır.
Klinik araştırmalardaki sıklıkları ender olmakla birlikte, majör ya da şiddetli kanamalar oluşabilir ve lokasyondan bağımsız olarak, engellilik oluşturucu, yaşamı tehdit edici, hatta ölümcül sonuçlara yol açabilir.
Elektif total kalça ya da diz replasman cerrahisi geçiren erişkin hastalarda venöz tromboembolik olayların önlenmesi: Bütünsel kanama oranları, tedavi grupları arasında benzer nitelikteydi ve anlamlı farklılık göstermemekteydi.
Bir ya da daha fazla risk faktörüne sahip, non-valvuler atriyal fibrilasyonlu erişkin hastalarda inme ve sistemik embolizmin önlenmesi: Majör kanama aşağıdaki kriterlerden bir ya da daha fazlasını karşılamaktaydı:
• Hemoglobinde en az litrede 20 g azalma ile birlikte olan, ya da en az 2 ünite kan ya da hücre konsantresi transfüzyonu ile sonuçlanan kanama;
• Kritik bir alanda ya da organda semptomatik kanama; kompartman sendromuyla birlikte intraoküler, intrakraniyel, intraspinal ya da intramusküler; retroperitoneal kanama, intra-artiküler kanama ya da perikardiyal kanama.
Majör kanamalar aşağıdaki kriterlerden bir ya da daha fazlasını karşıladığında yaşamı tehdit edici olarak sınıflandırıldı:
• Ölümcül kanama; semptomatik intrakraniyel kanama; hemoglobinde en az litrede 50 g azalma; en az 4 ünite kan ya da hücre konsantresi transfüzyonu; intravenöz inotropik ajanların kullanılmasını gerektiren hipotansiyon ile birlikte olan kanama; cerrahi girişim gerektiren kanama.
Günde iki kez 110 mg ya da 150 mg dabigatran eteksilat almak üzere randomize edilen olgularda yaşamı tehdit edici kanama, hemorajik inme ve intrakraniyel kanama riski, varfarine kıyasla anlamlı olarak daha düşüktü [p <0.05]. Her iki dabigatran eteksilat doz büyüklüğünde aynı zamanda, total kanama oranı da istatistiksel olarak anlamlı ölçüde daha düşüktü. Günde iki kez 110 mg dabigatran eteksilata randomize edilen olgularda majör kanama riski, varfarine kıyasla anlamlı olarak daha düşüktü (tehlike oranı 0.79, p = 0.0021).
Yan etkiler:
Kontrollü tüm çalışmalarda popülasyon başına herhangi bir tedavi grubunda bildirilen advers reaksiyonlar, aşağıdaki listelerde sistem organ sınıfı ve MedDRA tercihli terimlerine göre sınıflandırılmış olarak gösterilmektedir. Liste 1’de, tanımlanmış ve her iki endikasyon için de geçerli yan etkiler listelenmiştir. Liste 2’de, tanımlanan endikasyon spesifik yan etkiler ile birlikte, her iki endikasyonda da görülen ancak sıklık dereceleri farklı olan yan etkiler listelenmiştir.
Yan etkiler genellikle dabigatran eteksilatın farmakolojik etki mekanizmasıyla ilişkilidir ve değişik anatomik bölgeler ve organlarda ortaya çıkabilecek kanama-ilişkili olayları temsil etmektedir.
Kalça ya da diz replasman cerrahisinden sonra VTE’nin önlenmesi için tedavi edilen hastalarda dabigatran eteksilat ile gözlenen yan etkilerin insidansları, enoksaparin sınırları içindeydi.
İnmenin önlenmesi için tedavi edilen atriyal fibrilasyonlu hastalarda dabigatran eteksilat ile gözlenen yan etkilerin insidansları, dabigatran eteksilat kollarında daha yüksek bir oranda ortaya çıkan gastrointestinal bozuklukların dışında, varfarin sınırları içindeydi.
Belirtilen istenmeyen etkilerin sıklık oranları şu şekildedir:
Çok yaygın (> 1/10); yaygın (> 1/100 ila <1/10); yaygın olmayan (> 1/1.000 ila <1/100); seyrek (> 1/10.000 ila <1/1.000); çok seyrek (<1/10,000), bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor).
Liste 1: Elektif total kalça ya da diz replasman cerrahisinden sonra VTE’den primer korunma programı ve bir ya da daha fazla risk faktörüne sahip, non-valvuler atriyal fibrilasyonlu erişkin hastalarda tromboembolik inme ve sistemik embolizmin önlenmesi programında tanımlanan yan etkiler:
Kan ve lenf sistemi hastalıkları
Yaygın: Anemi
Yaygın olmayan: Trombositopeni
Bağışıklık sistemi hastalıkları
Yaygın olmayan: İlaç aşırı duyarlılığı, kaşıntı, döküntü Seyrek: Ürtiker
Sinir sistemi hastalıkları
Yaygın olmayan: İntrakraniyel hemoraji
Vasküler hastalıklar
Yaygın olmayan: Hematom, hemoraji
Solunum, göğüs bozuklukları ve mediyastinal hastalıklar
Yaygın: Epistaksis
Gastro-intestinal hastalıklar
Yaygın: Gastrointestinal hemoraji, abdominal ağrı, diyare, dispepsi, bulantı
Yaygın olmayan: Rektal hemoraji, hemoroidal hemoraji, gastrointestinal ülser, gastroözofajit, gastroözofajiyal reflü hastalığı, kusma, disfaji
Hepato-biliyer hastalıklar
Yaygın olmayan: Alanin aminotransferaz artışı, aspartat aminotransferaz artışı, karaciğer fonksiyonlarında anormallik/karaciğer fonksiyon testlerinde anormallik
Deri ve derialtı dokusu hastalıkları
Yaygın olmayan: Deride hemoraji
Böbrek ve idrar hastalıkları
Yaygın olmayan: Hematüri
Genel bozukluklar ve uygulama bölgesine ilişkin hastalıklar
Seyrek: Enjeksiyon bölgesinde hemoraji, kateter yerinde hemoraji
Yaralanma, zehirlenme ve prosedürel komplikasyonlar
Seyrek: İnsizyon bölgesinde hemoraji
Liste 2: Endikasyona spesifik olarak tanımlanan, ya da sıklık derecesi farklı olan yan etkiler:
Elektif total kalça ya da diz replasman cerrahisi geçiren hastalarda venöz tromboembolik olayların önlenmesi:
Kan ve lenf sistemi hastalıkları
Yaygın: Hemoglobinde azalma
Yaygın olmayan: Hematokritte azalma
Bağışıklık sistemi hastalıkları
Bilinmiyor: Bronkospazm
Vasküler hastalıklar
Yaygın olmayan: Yarada hemoraji
Hepato-biliyer hastalıklar
Yaygın olmayan: Karaciğer enzimlerinde artış, transaminazlarda artış, hiperbilirübinemi
Kas-iskelet, bağ dokusu ve kemik hastalıkları
Yaygın olmayan: Hemartroz
Genel bozukluklar ve uygulama bölgesine ilişkin hastalıklar
Yaygın olmayan: Kanlı akıntı
Yaralanma, zehirlenme ve prosedürel komplikasyonlar
Yaygın olmayan: Travmatik hemoraji, post-prosedürel hematom, post-prosedürel hemoraji, post-operatif anemi, post-prosedürel akıntı, yarada sekresyon
Cerrahi ve tıbbi prosedürler
Yaygın olmayan: Yara drenajı Seyrek: Post-prosedürel drenaj
Bir ya da daha fazla risk faktörüne sahip, non-valvuler atriyal fibrilasyonlu erişkin hastalarda inme ve sistemik embolizmin önlenmesi:
Kan ve lenf sistemi hastalıkları
Yaygın olmayan: Hemoglobinde azalma
Seyrek:
Hematokritte azalma
Bağışıklık sistemi hastalıkları
Çok seyrek: Bronkospazm
Solunum, göğüs bozuklukları ve mediyastinal hastalıklar
Yaygın olmayan: Hemoptizi
Seyrek:
Seyrek:
Yaygın:
Hepato-biliyer hastalıklar
Karaciğer enzimlerinde artış, hiperbilirübinemi
Kas-iskelet, bağ dokusu ve kemik hastalıkları
Hemartroz
Böbrek ve idrar hastalıkları
4.9. Doz aşımı ve tedavisi
PRADAXA® uygulamasını izleyen doz aşımı, farmakodinamik özelliklerine bağlı olarak hemorajik komplikasyonlara yol açabilir. PRADAXA®’nın farmakodinamik etkisini antagonize edecek spesifik bir antidot bulunmamaktadır. Önerilenlerin ötesinde PRADAXA® dozları hastada kanama riskinin artmasına neden olur. Aşırı antikoagülasyon PRADAXA®’ya son verilmesini gerektirebilir. Hemorajik komplikasy onlar durumunda, tedaviye son verilmeli ve kanamanın kaynağı araştırılmalıdır. Dabigatran esas olarak böbrek yoluyla atıldığı için, yeterli bir diürez sağlanmalıdır.
Uygun bir standart tedavi, örn. endikasyona göre cerrahi hemostaz ve kan hacmi replasmanı yapılmalıdır. Ek olarak, taze tam kan ya da taze donmuş plazma kullanılması da düşünülebilir. Proteine bağlanma düşük olduğu için, dabigatran diyalize edilebilir, ancak bu klinik zeminde diyaliz kullanımı konusundaki klinik deneyim kısıtlıdır.
Aktive protrombin kompleksi konsantreleri (örn. FEIBA) ya da rekombinant Faktör VIIa, veya koagülasyon faktörleri II, IX ya da X konsantreleri gündeme getirilebilir. Bu ajanların dabigatranın antikoagülan etkisinin tersine çevrilmesindeki rolünü destekleyen bazı deneysel veriler bulunmaktadır, ancak klinik ortamlardaki yararları henüz sistematik bir şekilde ortaya konulmamıştır. Trombositopeninin bulunduğu ya da uzun etkili anti-trombositer ilaçların kullanılmış olduğu durumlarda aynı zamanda, trombosit konsantrelerinin kullanılması da düşünülmelidir. Doktorun muhakemesi doğrultusunda her türlü semptomatik tedavi uygulanmalıdır.
5. FARMAKOLOJİK ÖZELLİKLER
5.1. Farmakodinamik özellikler
Farmakoterapötik grup: Oral direkt trombin inhibitörü ATC Kodu: B01A E07 - dabigatran eteksilat
Etki mekanizması:
Dabigatran eteksilat, herhangi bir farmakolojik aktivite göstermeyen, küçük moleküllü bir ön ilaçtır. Oral uygulamadan sonra, dabigatran eteksilat hızla absorbe edilir ve, plazma ile karaciğerde esteraz katalizli hidroliz reaksiyonu yoluyla dabigatran haline dönüştürülür. Dabigatran güçlü, yarışmacı, geri dönüşümlü, direkt bir trombin inhibitörüdür ve plazmada bulunan esas aktif maddedir.
Trombin (serin proteaz) koagülasyon süreci sırasında fibrinojenin fibrin haline dönüşmesini sağladığı için, bu maddenin inhibisyonu trombüs oluşmasını engeller. Dabigatran aynı zamanda, serbest trombin, fibrine bağlı trombin ve trombin-indüksiyonlu trombosit agregasyonunu da inhibe etmektedir.
In-vivo ve eks-vivo hayvan çalışmalarında, çeşitli hayvan tromboz modellerinde, intravenöz uygulamadan sonra dabigatranın ve oral uygulamadan sonra dabigatran eteksilatın antitrombotik etkinlik ve antikoagülan aktivite gösterdikleri ortaya konulmuştur.
Plazma dabigatran konsantrasyonları ve antikoagülan etkinin derecesi arasında yakın bir bağıntı vardır. Dabigatran aktive parsiyel tromboplastin zamanını (aPTT) uzatır.
Majör eklem replasman cerrahisinden sonra VTE profilaksisi klinik araştırmaları:
İki büyük, randomize, paralel gruplu, çift-kör, doz doğrulama araştırmasında (biri diz replasman cerrahisi ve biri kalça replasman cerrahisi), elektif majör ortopedik cerrahi uygulanan hastaların bir bölümüne cerrahiden sonraki 1-4 saat içinde hemostazın sağlanmış olması kaydıyla 75 mg ya da 110 mg dabigatran eteksilat verildi ve bunu daha sonra, günde bir kez 150 ya da 220 mg’lık dozlar izledi; diğer hastalara ise cerrahi girişim gününde, cerrahi uygulamadan önce, ve daha sonra günde bir kez 40 mg enoksaparin uygulandı.
Tedaviye RE-MODEL araştırmasında (diz replasmanı) 6 - 10 gün, RE-NOVATE araştırmasında (kalça replasmanı) 28 - 35 gün devam edildi. Toplam olarak, sırasıyla 2076 (diz) ve 3494 (kalça) hasta tedavi edildi.
Diz çalışmasının (RE-MODEL) primer sonlanım noktasına yönelik sonuçları (asemptomatik venöz tromboembolizm (VTE) dahil olmak üzere total VTE, artı tüm nedenlere bağlı mortalite), her iki dabigatran eteksilat dozundaki antitrombotik etkinin, enoksaparinin etkisinden istatistiksel olarak daha aşağıda olmadığını göstermekteydi.
Benzer şekilde, asemptomatik VTE dahil total VTE ve tüm nedenlere bağlı mortalite, kalça çalışmasında da (RE-NOVATE) primer sonlanım noktasını oluşturmaktaydı. Burada da günde
bir kez verilen her iki dabigatran eteksilat dozu, günde 40 mg enoksaparinden istatistiksel olarak daha aşağıda değildi.
Başka üçüncü bir randomize, paralel gruplu, çift-kör araştırmada (RE-MOBILIZE), elektif total diz cerrahisi geçiren hastalara, cerrahiden sonraki 6-12 saat içinde 75 mg ya da 110 mg dabigatran eteksilat verildi ve bunu daha sonra günde bir kez 150 ya da 220 mg’lık dozlar izledi. Tedavi süresi 12-15 gündü. Toplam 2615 hasta randomize edildi ve 2596’sı tedavi edildi. Enoksaparinin karşılaştırma dozu, ABD ürün bilgisi doğrultusunda günde iki kez 30 mg’dı. RE-MOBILIZE araştırmasında ’aşağıda olmayış’ gösterilmedi. Karşılaştırılan ajanlar arasında kanama yönüyle istatistiksel farklılık bulunmamaktaydı.
Ayrıca, Japon hastalara elektif total diz replasman cerrahisinden sonraki gün 110 mg, 150 mg ya da 220 mg dozlarında dabigatran eteksilat uygulanan, randomize, paralel gruplu, çift-kör, plasebo kontrollü faz II çalışmasının sonuçları değerlendirildi. Bu Japon çalışmasında, dabigatran eteksilatın etkinliği için net bir doz-yanıt bağıntısı ve plaseboya benzer bir kanama profili bulundu.
RE-MODEL ve RENOVATE araştırmalarında, ilgili çalışma ilacına randomizasyon cerrahi girişimden önce, ve RE-MOBILIZE ve Japon araştırmalarında ise cerrahi girişimden sonra yapılmıştı. Bu konu, özellikle araştırmaların güvenlilik değerlendirmeleri yapılırken dikkate alınması gereken bir noktadır. Bu nedenle, Tablo 1’de araştırmalar, cerrahiden önce ve cerrahiden sonra randomizasyon yapılan araştırmalar şeklinde gruplandırılmıştır.
Majör VTE ve VTE-ilişkili mortalite sonlanım noktası ve belirlenen majör kanama sonlanım noktalarına ilişkin veriler aşağıda Tablo 1’de gösterilmektedir. VTE, derin ven trombozu ve pulmoner embolizmin bileşik insidansı olarak tanımlanmıştı.
Tablo 1: RE-MODEL ve RE-NOVATE ortopedik cerrahi çalışmalarının tedavi dönemlerinde majör VTE ve VTE-ilişkili mortalite analizleri
Araştırma | Dabigatran eteksilat 220 mg | Dabigatran eteksilat 150 mg | Enoksaparin 40 mg |
RE-NOVATE (kalça) 1 | |||
N | 909 | 888 | 917 |
İnsidans (%) | 28 (3.1) | 38 (4.3) | 36 (3.9) |
Enoksaparine kıyasla risk farklılığı (%) | - 0.8 | 0.4 | |
%95 GA | - 2.5, 0.8 | - 1.5, 2.2 | |
Enoksaparine kıyasla risk oranı | 0.78 | 1.09 | |
%95 GA | 0.48, 1.27 | 0.70, 1.70 | |
RE-MODEL (diz) 1 | |||
N | 506 | 527 | 511 |
İnsidans (%) | 13 (2.6) | 20 (3.8) | 18 (3.5) |
Enoksaparine kıyasla risk farklılığı (%) | -1.0 | 0.3 | |
%95 GA | -3.1, 1.2 | -2.0, 2.6 | |
Enoksaparine kıyasla | 0.73 | 1.08 | |
risk oranı | |||
%95 GA | 0.36, 1.47 | 0.58, 2.01 | |
RE-MOBILIZE (diz)2 | Enoksaparin 60 mg | ||
N | 618 | 656 | 668 |
İnsidans (%) | 21 (3.4) | 20 (3.0) | 15 (2.2) |
Enoksaparine kıyasla risk farklılığı (%) | 1.2 | 0.8 | |
%95 GA | (-0.7, 3.0) | (-0.9, 2.5) | |
Enoksaparine kıyasla risk oranı | 1.51 | 1.36 | |
%95 GA | (0.79, 2.91) | (0.70, 2.63) | |
Japon diz çalışması | |||
Plasebo | |||
N | 102 | 113 | 104 |
İnsidans (%) | 0 | 2 (1.8) | 6 (5.8) |
Plaseboya kıyasla risk farklılığı (%) | -5.8 | -4.0 | |
%95 GA | (-10.3, -1.3) | (-9.1, 1.1) | |
1 Cerrahi öncesi randomizasyon çalışmaları 2 Cerrahi sonrası randomizasyon çalışmaları | |||
Bir ya da daha fazla risk faktörüne sahip, non-valvuler atriyal fibrilasyonlu erişkin hastalarda inme ve sistemik embolizmin önlenmesi klinik araştırmaları:
Dabigatran eteksilatın etkinliğine yönelik klinik kanıtlar RE-LY (Uzun dönemli antikoagülan tedavinin randomize değerlendirmesi / Randomized Evaluation of Long-term anticoagulant therapY) çalışmasından gelmektedir. Bu çalışma, orta ile yüksek derecede inme ya da sistemik embolizm riski taşıyan atriyal fibrilasyonlu hastalarda körlemeli iki dabigatran eteksilat dozu (110 mg bid ve 150 mg bid) ile açık tasarımlı varfarinin karşılaştırıldığı, çok-merkezli, çokuluslu, randomize, paralel gruplu bir çalışmaydı. Bu çalışmadaki primer amaç, bileşik sonlanım noktası olan inme ve sistemik embolik olayların (SEO) ortaya çıkışını azaltmada dabigatranın varfarinden aşağıda olup olmadığının belirlenmesiydi.
RE-LY çalışmasında, ortalama yaşı 71.5 ve ortalama CHADS2 skoru 2.1 olan, toplam 18,113 hasta tandomize edildi. Popülasyonda CHADS2 skoru 1, 2 ve >3 olan hastaların oranları hemen hemen eşitti. Hasta popülasyonunun %64’ü erkek, %70’i beyaz, ve %16’sı Asyalıydı. RE-LY çalışmasında medyan tedavi süresi 20 aydı ve dabigatran eteksilat, koagülasyon izlemesi yapılmaksızın, sabit doz şeklinde verildi. Belgelendirilmiş non-valvular atriyal fibrilasyon (AF), örn. ısrarlı ya da paroksismal AF’ye ek olarak, hastalarda aşağıdaki inmeye yönelik ek risk faktörlerinden biri de bulunmaktaydı:
• Önceden geçirilmiş inme, geçici iskemik atak ya da sistemik embolizm
• Sol ventrikül ejeksiyon fraksiyonu < %40
• Semptomatik kalp yetmezliği, > NYHA Sınıf 2
• Yaş > 75
• Yaş > 65 ve birlikte şu tablolardan biri; diabetes mellitus, koroner arter hastalığı, ya da hipertansiyon
Bu araştırmada hastalardaki eşzamanlı hastalıklar, hipertansiyon %79, diyabet %23 ve KAH %28 şeklindeydi. Hasta popülasyonunun %50’si, yaşam boyu total temas 2 aydan daha kısa şeklinde tanımlanmak üzere, VKA naif hastalardı. Popülasyonun %32’si hiç VKA kullanmamıştı. Varfarine randomize olan hastalar için, araştırma süresinde terapötik aralık (INR 2 ile 3) içinde olunan zaman, medyan %67 idi. Eşzamanlı alınan ilaçlar arasında aspirin (olguların %25’i çalışma süresinin en az %50’sinde kullandı), klopidogrel (%3.6), ASA+klopidogrel (%2), NSAEİ (%6.3), beta blokerler (%63.4), diüretikler (%53.9), statinler (%46.4), ADE inhibitörleri (%44.6), anjiyotensin reseptör blokerleri (%26.1), oral hipoglisemikler (%17.5), insülin (%5.2), digoksin (%29.4), amiodaron (%11.3), diltiazem (%8.9), verapamil (%5.4), ve proton pompası inhibitörleri (%17.8) bulunuyordu.
Primer sonlanım noktası olan inme ve sistemik embolizm için, varfarine kıyasla farklı bir risk oranına sahip alt-gruplar (örn. yaş, ağırlık, cinsiyet, böbrek fonksiyonu, etnisite vb.) tanımlanmadı.
Bu çalışmada, günde iki kez 110 mg dozunda dabigatran eteksilatın, atriyal fibrilasyonlu hastalarda inme ve sistemik embolizmin önlenmesinde varfarinden aşağıda olmadığı ve intrakraniyel hemoraji ve total kanama riskinin daha az olduğu ortaya konuldu. Yüksek doz olan günde iki kez 150 mg, iskemik ve hemorajik inme, vasküler ölüm, intrakraniyel hemoraji ve total kanama riskini varfarine kıyasla anlamlı olarak azaltmaktadır. Düşük dabigatran dozu, varfarine kıyasla anlamlı olarak daha düşük bir majör kanama riskine sahiptir.
Kilit önem taşıyan sonuçlar, Şekil 1 ve Tablo 2-6’da verilmektedir:
Tablo 2: RE-LY’da çalışma dönemi boyunca ilk inme ya da SEO oluşumunun (primer sonlanım noktası) analizi
Dabigatran eteksilat 150 mg bid | Dabigatran eteksilat 110 mg bid | Varfarin | |
Randomize edilen olgular | 6076 | 6015 | 6022 |
İnme ve/veya SEO | |||
İnsidanslar (%) | 134 (1.11) | 183 (1.54) | 202 (1.71) |
Varfarine kıyasla tehlike oranı (%95 GA) | 0.65 (0.52, 0.81) | 0.90 (0.74, 1.10) | |
p değeri üstünlüğü | p = 0.0001 | p = 0.2943 |
%’ler yıllık olay oranıdır.
Şekil 1: İlk inme ya da sistemik embolizme kadar geçen zamanın Kaplan-Meyer tahmin eğrileri
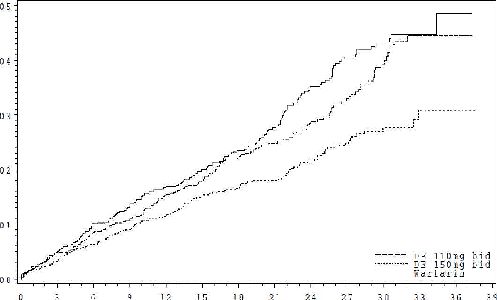
Randomizasyondan itibaren geçen zaman (ay)
Tablo 3: RE-LY’da çalışma dönemi boyunca ilk iskemik ya da hemorajik inme oluşumunun analizi
Dabigatran eteksilat 150 mg bid | Dabigatran eteksilat 110 mg bid | Varfarin | |
Randomize edilen olgular | 6076 | 6015 | 6022 |
İnme | |||
İnsidanslar (%) | 122 (1.01) | 171 (1.44) | 186 (1.58) |
Varfarine kıyasla tehlike oranı (%95 GA) | 0.64 (0.51, 0.81) | 0.91 (0.74, 1.12) | |
p değeri | 0.0001 | 0.3828 | |
SEO | |||
İnsidanslar (%) | 13 (0.11) | 15 (0.13) | 21 (0.18) |
Varfarine kıyasla tehlike oranı (%95 GA) | 0.61 (0.30, 1.21) | 0.71 (0.37, 1.38) | |
p değeri | 0.1582 | 0.3099 | |
İskemik inme | |||
İnsidanslar (%) | 103 (0.86) | 152 (1.28) | 134 (1.14) |
Varfarine kıyasla tehlike oranı (%95 GA) | 0.75 (0.58, 0.97) | 1.13 (0.89, 1.42) | |
p değeri | 0.0296 | 0.3139 | |
Hemorajik inme | |||
İnsidanslar (%) | 12 (0.10) | 14 (0.12) | 45 (0.38) |
Varfarine kıyasla tehlike oranı (%95 GA) | 0.26 (0.14, 0.49) | 0.31 (0.17, 0.56) | |
p değeri | <0.001 | <0.001 |
%’ler yıllık olay oranıdır.
Tablo 4: RE-LY’da çalışma dönemi boyunca tüm nedenlere bağlı ya da kardiyovasküler sağkalım analizi
Dabigatran eteksilat 150 mg bid | Dabigatran eteksilat 110 mg bid | Varfarin | |
Randomize edilen olgular | 6076 | 6015 | 6022 |
Tüm nedenlere bağlı mortalite | |||
İnsidanslar (%) | 438 (3.64) | 446 (3.75) | 487 (4.13) |
Varfarine kıyasla tehlike oranı (%95 GA) | 0.88 (0.77, 1.00) | 0.91 (0.80, 1.03) | |
p değeri | 0.0517 | 0.1308 | |
Vasküler mortalite | |||
İnsidanslar (%) | 274 (2.28) | 289 (2.43) | 317 (2.69) |
Varfarine kıyasla tehlike oranı (%95 GA) | 0.85 (0.72, 0.99) | 0.90 (0.77, 1.06) | |
p değeri | 0.0430 | 0.2081 |
%’ler yıllık olay oranıdır.
İnme, sistemik embolizm, pulmoner embolizm, akut miyokard enfarktüsü, vasküler ölümler ve majör kanamalardan oluşan ağırlıklandırılmamış bileşik klinik sonlanım noktasıyla ölçülen net klinik yarar (NKY) değerlendirilmiştir ve Tablo 7’nin bir bölümü şeklinde sunulmaktadır. Dabigatran eteksilat gruplarındaki yıllık olay oranları, varfarin grubuna kıyasla daha düşüktü. Bu bileşik sonlanım noktası için risk azalması, dabigatran eteksilat 110 mg bid ve 150 mg bid tedavi gruplarında %8 ve %10 oldu. İncelenen diğer bileşenler arasında bulunan tüm hospitalizasyonlar konusunda, dabigatran eteksilat 110 mg bid grubunda varfarine kıyasla anlamlı olarak daha az hospitalizasyon bulunmaktaydı (%7 risk azalması, %95 GA 0.87, 0.99, p = 0.021).
Tablo 5: Değerlendirilen diğer ölçümler
Dabigatran eteksilat 150 mg bid | Dabigatran eteksilat 110 mg bid | Varfarin | |
Randomize edilen olgular | 6076 | 6015 | 6022 |
İnme/SEO/Ölüm | |||
İnsidanslar (%) | 520 (4.32) | 577 (4.85) | 613 (5.20) |
Varfarine kıyasla tehlike oranı (%95 GA) | 0.83 (0.74, 0.93) | 0.93 (0.83, 1.045) | |
p değeri | 0.0015 | 0.2206 | |
İnme/SEO/PE/ME/Ölüm/Maj ör kanama (net klinik yarar) | |||
İnsidanslar (%) | 848 (7.05) | 863 (7.25) | 925 (7.84) |
Varfarine kıyasla tehlike oranı (%95 GA) | 0.90 (0.82, 0.99) | 0.92 (0.84, 1.01) | |
p değeri | 0.0254 | 0.0852 | |
Pulmoner embolizm | |||
İnsidanslar (%) | 18 (0.15) | 14 (0.12) | 12 (0.10) |
Varfarine kıyasla tehlike | 1.41 (0.71, 3.06) | 1.16 (0.54, 2.51) |
oranı (%95 GA) | |||
p değeri | 0.2980 | 0.7076 | |
Miyokard enfarktüsü (sessiz enfarktüs dahil) | |||
İnsidanslar (%) | 97 (0.81) | 98 (0.82) | 75 (0.64) |
Varfarine kıyasla tehlike oranı (%95 GA) | 1.27 (0.94, 1.71) | 1.29 (0.96, 1.75) | |
p değeri | 0.1240 | 0.0929 |
Tablo 6: Karaciğer fonksiyon testleri
RE-LY çalışmasında karaciğer fonksiyon testlerindeki potansiyel anormallikler, dabigatran eteksilat ile tedavi edilen hastalarda, varfarin tedavisindeki hastalara kıyasla, karşılaştırılabilir ya da daha düşük bir insidansta ortaya çıkmıştır.
Dabigatran eteksilat | Dabigatran eteksilat | Varfarin N (%) | |||||||||||||||||||||||||||||||||||||||||||||
150 mg bid | 110 mg bid | ||||||||||||||||||||||||||||||||||||||||||||||
N (%) | N (%) | ||||||||||||||||||||||||||||||||||||||||||||||
Toplam tedavi edilen | 6059 (100.0) | 5983 (100.0) | 5998 (100.0) | ||||||||||||||||||||||||||||||||||||||||||||
ALT ya da AST >3 x NÜS | 106 (1.7) | 118 (2.0) | 125 (2.1) | ||||||||||||||||||||||||||||||||||||||||||||
ALT ya da AST >5 x NÜS | 45 (0.7) | 36 (0.6) | 50 (0.8) | ||||||||||||||||||||||||||||||||||||||||||||
ALT ya da AST >3 x NÜS + | 14 (0.2) | 11 (0.2) | 21 (0.4) | ||||||||||||||||||||||||||||||||||||||||||||
Bilirübin >2 x NÜS | 5.2. Farmakokinetik özelliklerGenel özellikler Sağlıklı gönüllülerde oral dabigatran eteksilat uygulamasından sonra, dabigatranın plazmadaki farmakokinetik profili, plazma konsantrasyonlarında hızlı bir artış, ve doruk konsantrasyona (Cmaks) uygulama sonrası 0.5 ve 2.0 saat içerisinde ulaşılmasıyla karakterizedir. Cmaks ve plazma konsantrasyon-zaman eğrisi altındaki alan (EAA) doz ile orantılıydı. Cmaks’tan sonra, dabigatran plazma konsantrasyonları bieksponansiyel azalma gösterir, ve ortalama terminal yarı ömür sağlıklı yaşlı olgularda yaklaşık 11 saattir. Tekrarlı dozlardan sonra, 12-14 saat civarında bir terminal yarı-ömür gözlenir. Yarı ömür dozdan bağımsızdır. Ancak, aşağıda Tablo 7’de gösterildiği gibi, böbrek fonksiyonları bozulmuş ise, yarı-ömür uzamaktadır. Tablo 7: Total dabigatranın sağlıklı olgular ve böbrek fonksiyonları bozulmuş olgulardaki yarı-ömrü
HMPC kapsülü şeklinde oral dabigatran eteksilat uygulamasından sonra dabigatranın mutlak biyoyararlanımı %6.5 civarındadır. Yiyecekler dabigatran eteksilatın biyoyararlanımını etkilemez, ama pik plazma konsantrasyonlarına ulaşma zamanını 2 saat süreyle geciktirir. Pelletler HPMC kapsül kabuğu olmaksızın alındığında, oral biyoyararlanım, referans kapsül formülasyonuna kıyasla %75 artabilir. Dolayısıyla klinik kullanımda, dabigatran eteksilat biyoyararlanımında arzu edilmeyen artışlardan kaçınmak için, HPMC kapsüllerin bütünlüğü her zaman için korunmalıdır. Bu nedenle hastalara kapsülleri açmamaları ve pelletleri tek başına almamaları (örn. yiyecek ve içeceklerin üzerine serpiştirme) öğütlenmelidir (bkz. "Pozoloji ve uygulama şekli"). Emilim: Dabigatran eteksilatın cerrahi girişimden 1-3 saat sonraki post-operatif absorpsiyonunun değerlendirildiği bir çalışmada, yüksek pik plazma konsantrasyonları içermeyen düz bir plazma konsantrasyon-zaman profili göstererek, sağlıklı gönüllülerdekine kıyasla daha yavaş bir absorpsiyonu olduğu ortaya konulmuştur. Pik plazma konsantrasyonlarına uygulamadan 6 saat sonra, ya da cerrahiyi izleyen 7 ile 9 saat sonra ulaşılmıştır (BISTRO Ib). Ancak anestezi, gastrointestinal parezi ve cerrahi etkiler gibi katkıda bulunan faktörler dolayısıyla, hastaların bir bölümünde oral ilaç formülasyonundan bağımsız olarak, absorpsiyon gecikmeleri yaşanabileceği kaydedilmiştir. Bu çalışmada absorpsiyon bozukluğunun sonraki dozlarda kalıcı olup olmadığı ele alınmamış olmakla birlikte, sonraki bir çalışmada, yavaş ve gecikmiş absorpsiyonun genellikle sadece cerrahi girişimin yapıldığı gün söz konusu olduğu gösterilmiştir. Daha sonraki günlerde dabigatran absorpsiyonu hızlı olmuş ve pik plazma konsantrasyonlarına ilaç uygulamasından 2 saat sonra ulaşılmıştır. Dağılım: Dabigatranın insan plazma proteinlerine konsantrasyondan bağımsız olarak, düşük oranda (%34-35) bağlandığı gözlenmiştir. Dabigatranın 60 - 70 L düzeyindeki dağılım hacmi, total vücut sıvısını aşar; bu durum dabigatranın dokulara orta derecede dağıldığını göstermektedir. Biy otransformasy on: Oral uygulamadan sonra, dabigatran eteksilat hızla ve tamamen, plazmadaki aktif form olan dabigatran haline dönüştürülür. Ön-ilaç dabigatran eteksilatın esteraz katalizli hidroliz reaksiyonu yoluyla aktif madde dabigatrana parçalanması, başlıca metabolik reaksiyondur. Dabigatran konjügasyona maruz kalarak, farmakolojik olarak aktif açilglukuronidler oluşturur. Dört pozisyonel izomer, 1-O, 2-O, 3-O, 4-O-açilglukuronid vardır, ve her biri plazmadaki total dabigatranın %10’undan azını oluşturur. Eser miktarlardaki diğer metabolitler, sadece son derecede hassas analitik metodlar ile saptanabilir niteliktedir. Eliminasyon: Dabigatran metabolizması ve ekskresyonu, sağlıklı erkek olgularda tek intravenöz radyoetiketli dabigatran dozunu izleyerek incelenmiştir. Tek intravenöz dozdan sonra, dabigatran kökenli radyoaktivite esas olarak idrar ile elimine edilir (%85). Fekal ekskresyon uygulanan dozun %6’sından sorumludur. Uygulama sonrası 168. saatte geri kazanılan total radyoaktivite, uygulanan dozun %88 - 94’ü aralığındadır. Dabigatran esas olarak idrarla değişmemiş halde elimine edilir; eliminasyon hızı glomerüler filtrasyon hızına karşılık gelecek şekilde, yaklaşık 100 ml/dk’dır. Doğrusallık/Doğrusal olmayan durum: Dabigatran doğrusal bir kinetik gösterir. Hastalaı daki karakteristik özellikler Yaş: Pediyatrik popülasyon: Dabigatran eteksilat 18 yaşın altındaki hastalarda incelenmemiştir. Çocuklarda dabigatran eteksilat kullanımı önerilmemektedir. Geriyatrik popülasyon: Faz 1 çalışmalarında yaşlı olgularda yürütülen spesifik farmakokinetik çalışmalarda, genç olgulara kıyasla EAA’da %40 ile 60, ve Cmaks’ta %25’in üzerinde bir artış olduğu gösterilmiştir. Yaşlı (>65) erkek ve kadın olgularda EAAT,ss ve Cmaks,ss değerleri, yaşlı kadınlarda genç kadınlara kıyasla yaklaşık 1.9 ve 1.6 kat, yaşlı erkeklerde ise 18 - 40 yaşındaki erkek olgulardan yaklaşık 2.2 ve 2.0 kat daha yüksektir. Sistemik dabigatran temasında gözlenen artış, kreatinin klerensindeki yaşa bağlı azalma ile korelasyon göstermektedir. Yaşın sistemik dabigatran teması üzerindeki etkisi RE-LY çalışmasında doğrulanmıştır; 65 ile 75 yaş arasındaki olgulardakine kıyasla, çukur konsantrasyonlar, >75 yaşındaki olgularda %31 civarında yüksek, ve 65 yaşın altındaki olgularda %22 civarında daha düşük bulunmuştur. Cinsiyet: VTE’den primer korunma çalışmalarında kadın hastalardaki ilaç teması %40 ile %50 civarında daha yüksek bulunmuştur. Atriyal fibrilasyon hastalarında kadınlar ortalama olarak %30 daha yüksek çukur ve doz-sonrası konsantrasyonlara sahiptiler. Bu bulgu klinik önem taşımamaktadır. Irk: Dabigatranın farmakokinetik özellikleri Beyaz ve Japon gönüllülerde tek ve tekrarlı dozlardan sonra araştırılmıştır. Etnik köken dabigatranın farmakokinetik özelliklerini klinik önem taşıyacak şekilde etkilememektedir. Siyah hastalara ilişkin kısıtlı farmakokinetik veriler, önemli farklılık olmadığı izlenimini vermektedir. Vücut ağırlığı: Dabigatran çukur konsantrasyonları vücut ağırlığı >100 kg olan hastalarda, 50 - 100 kg olanlara kıyasla, %20 civarında daha düşüktür. Olguların büyük çoğunluğu (%80.8) >50 kg ile <100 kg kategorisi içindeydi ve belirgin bir farklılık saptanmamıştır. <50 kg olan hastalar için bulunan veriler kısıtlıdır. Böbrek yetmezliği: Bir Faz 1 çalışmasında, orta derecede böbrek yetmezliği (CrCL 30 - 50 ml/dk arasında) olan gönüllülerde oral dabigatran eteksilat uygulamasından sonraki dabigatran teması (EAA), böbrek yetmezliği olmayanlardakinden yaklaşık 3 kat daha yüksek bulunmuştur. Şiddetli böbrek yetmezliği (CrCL 10 - 30 ml/dk) olan az sayıdaki gönüllüde dabigatran teması (EAA), böbrek yetmezliği olmayan bir popülasyonda gözlenenden yaklaşık 6 kat daha yüksek ve yarı ömür 2 kat daha uzun bulunmuştur (bkz. Pozoloji ve uygulama şekli ve Kontrendikasy onlar). Karaciğer yetmezliği: Bir Faz 1 çalışmasında orta derecede karaciğer yetmezliği (Child Pugh B) olan 12 olguda, 12 kontrole kıyasla sistemik dabigatran temasında bir değişiklik görülmemiştir. Elektif total kalça ya da diz replasman cerrahisi geçiren erişkin hastalarda venöz tromboembolik olayların önlenmesi: Orta derecede ve şiddetli karaciğer yetmezliği (Child-Pugh sınıf B ve C) olan hastalar, ya da karaciğer enzimlerinde >2 Normal Üst Sınır (NÜS) yükselme görülen veya sağkalım üzerinde etkili olması beklenen bir karaciğer hastalığı olan hastalar klinik araştırmalara dahil edilmemiştir. Bir ya da daha fazla risk faktörüne sahip, non-valvuler atriyal fibrilasyonlu erişkin hastalarda inme ve sistemik embolizmin önlenmesi: Karaciğer enzimlerinde sürekli >2 Normal Üst Sınır (NÜS) yükselme görülen ya da hepatit A, B veya C dahil, ancak bunlarla sınırlı olmayan aktif karaciğer hastalığı bulunan hastalar klinik araştırmalara dahil edilmemiştir. Farmakokinetik etkileşimler In vitro etkileşim çalışmalarında sitokrom P450’de herhangi bir inhibisyon ya da indüksiyon görülmemiştir. Bu durum sağlıklı gönüllülerdeki in vivo çalışmalar ile doğrulanmıştır. Bu çalışmalarda dabigatran eteksilat ile aşağıdaki ilaçlar arasında herhangi bir etkileşim görülmemiştir; atorvastatin (CYP3A4) ve diklofenak (CYP2C9). Atorvastatin: Dabigatran eteksilat, bir CYP3A4 substratı olan atorvastatin ile birlikte uygulandığında, atorvastatin, atorvastatin metabolitleri ve dabigatranın sistemik temas düzeyleri değişmemişti. Bu durum arada etkileşim bulunmadığını göstermektedir. Diklofenak: Dabigatran eteksilat, bir CYP2C9 substratı olan diklofenak ile birlikte uygulandığında, her iki ilacın farmakokinetik özellikleri değişmemişti. Bu durum dabigatran eteksilat ve diklofenak arasında etkileşim bulunmadığını göstermektedir. P-gp inhibitörü / indükleyici etkileşimleri Dabigatran değil ama ön ilaç olan dabigatran eteksilat dışarı akış taşıyıcısı P-glikoproteinin (P-gp) bir subtratıdır. Bu nedenle, P-gp taşıyıcı inhibitörleri ve indükleyicileriyle birlikte uygulama araştırılmıştır. P-gp inhibitörleriyle birlikte uygulama Amiodaron: Dabigatran eteksilat oral tek doz 600 mg amiodaron ile birlikte uygulandığında, amiodaron ve aktif metaboliti DEA’nın absorpsiyon hızı ve boyutları esas olarak değişmemişti. Dabigatran EAA ve Cmaks değerleri sırasıyla yaklaşık %60 ve %50 oranında artmıştı. Verapamil: Dabigatran eteksilat oral verapamil ile birlikte uygulandığında, dabigatranın Cmaks ve EAA değerleri, verapamil dozunun uygulama zamanına ve formülasyonuna bağlı olarak artış göstermiştir. Dabigatran sistemik temasındaki en büyük artış, dabigatran eteksilatın alınmasından bir saat önce uygulanan çabuk salımlı verapamil formülasyonunun ilk dozunda gözlendi (%180 civarında Cmaks ve %150 civarında EAA artışları). Bu etki uzatılmış salımlı formülasyon uygulamasıyla (%90 civarında Cmaks ve %70 civarında EAA artışları) ya da tekrarlı verapamil doz uygulamasıyla (%60 civarında Cmaks ve %50 civarında EAA artışları) progresif bir şekilde azaldı. Bu durum, kronik verapamil tedavisinde, bağırsaktaki P-gp indüksiyonuyla açıklanabilir. Verapamil dabigatran eteksilattan 2 saat sonra verildiğinde anlamlı bir etkileşim gözlenmedi (%10 civarında Cmaks ve %20 civarında EAA artışları). Bu durum, dabigatran emiliminin 2 saat sonra tamamlanmasıyla açıklanmaktadır (bkz. Pozoloji ve uygulama şekli). Parenteral verapamil uygulamasına ilişkin veri bulunmamaktadır; etkileşimin mekanizması temelinde, anlamlı bir etkileşim beklenmemektedir. Ketokonazol: Ketokonazol, total dabigatran EAA0-» ve Cmaks değerlerini arttırmıştır. Bu artışlar tek doz 400 mg’dan sonra sırasıyla, %138 ve %135, tekrarlı 400 mg ketokonazol qd dozlarından sonra ise, sırasıyla, %153 ve %149 düzeyindeydi. Doruk konsantrasyon zamanı, terminal yarı-ömür ve ortalama kalış zamanları ketokonazol tarafından etkilenmemişti. Klaritromisin: Günde iki kez 500 mg klaritromisin dabigatran eteksilat ile birlikte uygulandığında, klinik önem taşıyan bir farmakokinetik etkileşim gözlenmemiştir (%15 civarında Cmaks ve %19 civarında EAA artışları). Kinidin: Kinidin, total 1000 mg dozuna kadar, iki saatte bir verilen 200 mg’lık dozlar şeklinde uygulandı. Dabigatran eteksilat bid ardışık 3 gün süreyle uygulandı ve 3. gün kinidin ile birlikte ya da kinidin olmaksızın verildi. Eşzamanlı kinidin ile, dabigatran EAAT,ss ve Cmaks,ss değerleri, sırasıyla ortalama %53 ve %56 artış gösterdi. P-gp substratlarıyla birlikte uygulama Digoksin: Dabigatran eteksilat, bir P-gp substratı olan digoksin ile birlikte uygulandığında, herhangi bir farmakokinetik etkileşim gözlenmemiştir. Ne dabigatran ne de ön-ilaç dabigatran eteksilat, klinik önem taşıyan bir P-gp inhibitörüdür. P-gp indükleyicileriyle birlikte uygulama Rifampisin: Önceden 7 gün süreyle 600 mg qd dozunda prob indükleyici rifampisin uygulaması, total dabigatran doruk ve total sistemik temasını, sırasıyla %65.5 ve %67 oranında azaltmıştır. İndükleyici etki daha sonra azaldı ve rifampisin tedavisinin kesilmesinden sonraki 7. günde, referansa yaklaştı. İkinci bir 7 günden sonra, biyoyararlanımda başka artış gözlenmedi. Trombosit inhibitörleriyle birlikte uygulama Asetilsalisilik asit (ASA): Dabigatran eteksilat ve asetilsalisilik asidin (ASA) birlikte uygulanmalarının kanama riski üzerindeki etkisi, atriyal fibrilasyonlu hastalara randomize yöntemle ASA uygulanan bir faz II çalışmada incelenmiştir. Lojistik regresyon analizi temelinde, ASA ve günde iki kez 150 mg dabigatran eteksilat uygulaması, herhangi bir kanama riskini, 81 mg ve 325 mg ASA ile %12’den, sırasıyla %18 ve %24’e yükseltebilir. Faz III RE-LY çalışmasında edinilen verilerden, 110 ya da 150 mg bid dabigatran eteksilat ile birlikte ASA ya da klopidogrel uygulamasının majör kanama riskini arttırabileceği gözlenmiştir. Ancak, birlikte ASA ya da klopidogrel uygulamasıyla kanama olayları oranındaki yükselme, aynı zamanda varfarin ile de gözlenmiştir. Kısa dönemli perioperatif analjezi için uygulanan NSAEİ’lerin, dabigatran eteksilat ile birlikte verildiklerinde, kanama riskinde artış ile ilişkili olmadıkları gösterilmiştir. Dabigatran eteksilat tedavisi sırasında, 12 saatten daha kısa yarı-ömürleri olan olağan NSAEİ kullanımı konusunda kısıtlı veriler bulunmaktadır, ve bu veriler ek kanama riski izlenimi oluşturmamıştır. Klopidogrel: Sağlıklı genç erkek gönüllüler üzerindeki bir faz I çalışmasında, dabigatran eteksilat ile birlikte klopidogrel kullanımı, kapiller kanama zamanlarında (KKZ), klopidogrel monoterapisine kıyasla daha fazla bir uzama ile sonuçlanmamıştı. Kombine tedavi ve ilişkili monoterapiler karşılaştırıldığında aynı zamanda, dabigatran EAAT,ss ve Cmaks,ss değerleri, dabigatran etkisinin koagülasyon ölçümleri olarak aPTT, ECT ya da TT (anti FIIa), ya da klopidogrel etkisinin ölçümü olarak trombosit agregasyonunun inhibisyonu (IPA) esas olarak değişmeksizin kalmıştı. 300 ya da 600 mg klopidogrel yükleme dozuyla, dabigatran EAAT,ss ve Cmaks,ss değerleri %30 ile 40 civarında yükseldi. Gastrik pH yükseltici ajanlarla birlikte uygulama Pantoprazol: Dabigatran eteksilat pantoprazol ile birlikte uygulandığında, dabigatran plazma konsantrasyon - zaman eğrisi altındaki alanda, yaklaşık %30 civarında bir azalma gözlenmiştir. Klinik araştırmalarda pantoprazol ve diğer proton-pompası inhibitörleri dabigatran eteksilat ile birlikte uygulanmış, ve kanama ya da etkinlik üzerinde bir etki gözlenmemiştir. Ranitidin: Dabigatran eteksilat ile birlikte ranitidin uygulamasının, dabigatran absorpsiyonu boyutları üzerinde klinik olarak anlamlı bir etkisi bulunmamaktaydı. 5.3. Klinik öncesi güvenlilik verileriAkut oral toksisite çalışmaları sıçanlar ve farelerde yürütülmüştür. Her iki türde de, tek oral uygulamadan sonraki yaklaşık letal doz, 2000 mg/kg’ın üzerindeydi. Köpekler ve Rhesus maymunlarında, oral yoldan 600 mg/kg dabigatran eteksilat uygulaması toksikolojik olarak anlamlı değişikliklere yol açmadı. Sıçanlarda maksimum 26 hafta ve Rhesus maymunlarında maksimum 52 haftaya kadar olan tekrarlı doz toksisite çalışmalarında, 300 mg/kg’a (serbest baz eşdeğeri) kadar dozlar kullanılmıştır. Genel olarak bu dozlar, gerek sıçanlar gerekse Rhesus maymunları tarafından dikkate değer ölçüde iyi tolere edilmiştir. Uygulamadan sonraki ilk 4 - 6 saat içinde travma ile ilişkili (örn. kan örneği alma) kanama problemleri gözlenmiştir ve doğrudan dabigatranın farmakodinamik aktivitesine bağlıdır. Teratoloji çalışmaları sıçanlar ve tavşanlarda 200 mg/kg’a (serbest baz eşdeğeri) kadar dozlar ile yürütülmüştür. Sıçanlarda 200 mg/kg (serbest baz eşdeğeri) dozunda, fetusların morfoj enezi üzerinde hafif bir etki gözlenmiştir. Tavşanlarda teratojenik etki kaydedilmemiştir. Sıçanlardaki fertilite çalışmasında, toksikolojik olarak kayda değer parental bulgular elde edilmemiştir. Yavrulara ilişkin parametreler yönüyle, 200 mg/kg (serbest baz eşdeğeri) doz grubunda, corpus luteumda hafif azalma ve pre-implantasyon kayıplarındaki artış, ortalama implantasyon sayısında azalmaya yol açmıştır. Kapsamlı in vitro ve in vivo çalışmalarda, mutajenik potansiyel belirtisi açığa çıkarılmamıştır. Sıçanlar ve farelerde yürütülen yaşam-boyu toksikoloji çalışmalarında, maksimum 200 mg/kg dabigatran dozlarına kadar (serbest baz eşdeğeri), tümörijenik potansiyel belirtisi bulunmamaktaydı. 6. FARMASÖTİK ÖZELLİKLER6.1. Yardımcı maddelerin listesiTartarik asit Arap zamkı Hipromelloz Dimetikon 350 Talk Hidroksipropil selüloz Carragenan Potasyum klorür Titanyum dioksit (E171) Gün batımı sarısı (E110) Indigo Carmin (E 132) Şellak N-bütil alkol İzopropil alkol Denatüre alkol Siyah demir oksit (E172) Saf su 6.2. GeçimsizliklerGeçerli değil. 6.3. Raf ömrü6.4. Saklamaya yönelik özel tedbirler6.5. Ambalajın niteliği ve içeriğiHer aluminyum blister şeridinde 10 sert kapsül. 6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemlerÖzel bir önlem gerekmemektedir. Kullanılmamış olan ürün ya da atık materyaller, ’Tıbbi Atıkların Kontrolü Yönetmeliği’ ve ’Ambalaj ve Ambalaj Atıklarının Kontrolü Yönetmelikleri’ne uygun olarak imha edilmelidir.
İLAÇ GENEL BİLGİLERİ
Boehringer Ingelheim İlaç Tic. A.Ş.
|




 Kanseri")
| Geri Ödeme Kodu | A12789 |
| Satış Fiyatı | 218.74 TL [ 5 Apr 2024 ] |
| Önceki Satış Fiyatı | 218.74 TL [ 1 Apr 2024 ] |
| Original / Jenerik | Original İlaç |
| Reçete Durumu | Normal Reçeteli bir ilaçdır. |
| Barkodu | 8699693150103 |
| Etkin Madde | Dabigatran |
| ATC Kodu | B01AE07 |
| Birim Miktar | 110 |
| Birim Cinsi | MG |
| Ambalaj Miktarı | 10 |
| Kan ve Kan Yapıcı Organlar > Antitrombotik İlaçlar > Dabigatran |
| İthal ( ref. ülke : Fransa ) ve Beşeri bir ilaçdır. |