OCREVUS 300 mg/ 10ml inf�zyonluk ��zelti haz�rlamak i�in konsantre (1 flakon) K�sa �r�n Bilgisi
{ Okrelizumab }
1. BE�ER� TIBB� �R�N�N ADI
OCREVUS 300 mg/10 mL inf�zyonluk ��zelti haz�rlamak i�in konsantre
2. KAL�TAT�F VE KANT�TAT�F B�LE��M
Etkin madde
Her flakon, 30 mg/mL konsantrasyonda ocrelizumab ��zeltisi i�erir. Toplamda her flakonda 10 mL'de 300 mg ocrelizumab bulunmaktad�r. Dil�syon sonras�nda elde edilen ila� konsantrasyonu yakla��k 1,2 mg/mL' dir.
Ocrelizumab, rekombinant DNA teknolojisiyle �in Hamsteri Yumurtal��� h�cre dizilerinde �retilen bir humanize monoklonal antikorudur.
Yard�mc� maddeler
Sodyum asetat trihidrat 21,4 mg Di�er yard�mc� maddeler i�in 6.1'e bak�n�z.
3. FARMAS�T�K FORMU
Steril
�nf�zyonluk ��zelti haz�rlamak i�in konsantre.
Berrak ila hafif opalesan ve renksiz ila a��k kahverengi ��zelti.
4. KL�N�K �ZELL�KLER
4.1. Terap�tik endikasyonlar
Ataklarla seyreden (RMS) veya primer progresif MS (PPMS) vakalar�nda endikedir.
4.2. Pozoloji ve uygulama �ekli
Tedavi, ciddi inf�zyonla ili�kili reaksiyonlar (��R) gibi �iddetli reaksiyonlar�n y�netiminde uygun t�bbi deste�e eri�imi olan, deneyimli bir sa�l�k uzman� taraf�ndan ba�lat�lmal� ve takip edilmelidir.
�nf�zyonla ili�kili reaksiyonlar i�in premedikasyon
��R'lerin s�kl��� ve �iddetini daha da azaltmak amac�yla her ocrelizumab inf�zyonundan �nce a�a��daki iki premedikasyon �nerilmektedir (bkz. B�l�m 4.4):
Her inf�zyondan yakla��k 30 dakika �nce 100 mg intraven�z metilprednisolon veya e�de�er dozda di�er kortikosteroidlerin uygulanmas�;
4.3. Kontrendikasyonlar
OCREVUS a�a��daki durumlarda kontrendikedir:
Ocrelizumaba veya B�l�m 6.1'de listelenen yard�mc� maddelerden herhangi birine kar�� a��r� duyarl�l��� olan hastalarda
4.4. �zel kullan�m uyar�lar� ve �nlemleri
�zlenebilirlik
Biyolojik t�bbi �r�nlerin izlenebilirli�ini art�rmak i�in, uygulanan �r�n�n ticari ad� ve seri numaras� net bir �ekilde hasta dosyas�na kaydedilmelidir.
�nf�zyonla �li�kili Reaksiyonlar (��R)
Ocrelizumab, sitokin sal�n�m�na ve/veya di�er kimyasal mediyat�rlerle ili�kili olabilecek inf�zyonla ili�kili reaksiyonlarla ili�kilendirilebilir.
��R semptomlar� herhangi bir ocrelizumab inf�zyonu s�ras�nda meydana gelebilir ancak s�kl�kla ilk inf�zyon s�ras�nda rapor edilmi�tir. ��R'ler inf�zyondan sonra 24 saat i�erisinde meydana gelebilir (bkz. B�l�m 4.8). Bu reaksiyonlar, ka��nt�, d�k�nt�, �rtiker, eritem, , bo�az irritasyonu, orofaringeal a�r�, dispne, faringeal veya laringeal �dem, y�zde k�zar�kl�k, hipotansiyon, y�ksek ate�, yorgunluk, ba� a�r�s�, sersemlik hali, bulant�,ta�ikardi ve anafilaksi �eklinde ortaya ��kabilir.
�nf�zyondan �nce:
�iddetli reaksiyonlar�n y�netimi: ��R, a��r� duyarl�l�k reaksiyonlar� ve/veya anafilaktik reaksiyonlar gibi �iddetli reaksiyonlar�n y�netimi i�in uygun kaynaklar haz�r bulundurulmal�d�r.
4.5. Di�er t�bbi �r�nler ile etkile�imler ve di�er etkile�im �ekilleri
Sitokrom P450 enzimleri, di�er metabolize edici enzimler veya ta��y�c�lar �zerinden hi�bir etkile�im beklenmedi�inden, etkile�im �al��malar� y�r�t�lmemi�tir.
A��lar
Ocrelizumab tedavisi sonras�nda canl� veya canl� aten�e a��lar ile ba����klaman�n g�venlili�i hen�z �al���lmam��t�r.
Ocrelizumab alan hastalarda tetanoz toksoid, 23-valansl� pn�mokoksal polisakkarit, keyhole limpet hemosiyanin neoantijen ve mevsimsel influenza a��lar�n�n etkilerine ait veri mevcuttur. (bkz. B�l�m 4.4 ve 5.1)
2 y�l� a�k�n tedavinin ard�ndan, S. Pn�moni, kabakulak, rubella ve varicella'ya kar�� pozitif antikor titresi olan hastalar�n oranlar�, ba�lang��taki ile ayn�d�r.
�mmunosupresif tedaviler:
Relapslar�n semptomatik tedavisi i�in kortikosteroidler d���nda ocrelizumab ile birlikte di�er immunosupresif tedavilerin kullan�lmas� �nerilmez (bkz. B�l�m 4.4).
�zel pop�lasyonlara ili�kin ek bilgiler:
Ocrelizumab ile herhangi bir farmakokinetik ila� etkile�imi �al��mas� yap�lmam��t�r.
Pediyatrik pop�lasyon:
Ocrelizumab ile pediyatrik pop�lasyonda herhangi bir farmakokinetik ila� etkile�imi �al��mas� yap�lmam��t�r.
4.6. Gebelik ve laktasyon
Gebelik kategorisi: C
�ocuk do�urma potansiyeli bulunan kad�nlar / Do�um kontrol� (Kontrasepsiyon) �ocuk do�urma potansiyeli bulunan kad�nlar, ocrelizumab al�rken ve son ocrelizumab inf�zyonundan sonra 12 ay boyunca do�um kontrol y�ntemi kullanmal�d�rlar (bkz. B�l�m 5.1 ve 5.2).
Gebelik d�nemi
Ocrelizumab�n gebe kad�nlarda kullan�m�na ili�kin yeterli veri mevcut de�ildir. Hayvanlar �zerinde yap�lan ara�t�rmalar �reme toksisitesinin bulundu�unu g�stermi�tir (bkz. B�l�m 5.3). �nsanlara y�nelik potansiyel risk bilinmemektedir.
Ocrelizumab�n gebe kad�nlarda kullan�m� ile ili�kili geli�imsel risk konusundaki veriler yetersizdir. Ocrelizumab bir imm�noglobulin G (IgG)'dir. IgG'nin plasenta bariyerini ge�ti�i bilinmektedir. Gebelikte ocrelizumaba maruz kalan annelerden do�an yenido�an ve bebeklerde canl� ya da canl� aten�e a��larla a��laman�n ertelenmesi d���n�lmelidir. Ocrelizumaba maruz kalan yenido�anlarda ve bebeklerde B-h�cre say�m� verisi toplanmam��t�r ve B-h�cre deplesyonun potansiyel devam s�resi bilinmemektedir (bkz. B�l�m 4.4).
Gebelik s�ras�nda di�er anti-CD20 antikorlara maruz kalm�� olan annelerin bebeklerinde ge�ici periferik B-h�cre deplesyonu ve lenfositopeni rapor edilmi�tir.
Hayvan �al��malar� (embriyo-fetal toksisite) teratojenik etkilere i�aret etmemektedir. Ancak, uteroda B-h�cre deplesyonu tespit edimi�tir. Do�um �ncesi ve sonras� geli�tirme �al��malar�nda �reme toksisitesi g�zlenmi�tir (bkz. B�l�m 5.3).
Anne i�in olan potansiyel fayda fet�se potansiyel riskten a��r basmad��� s�rece gebelikte ocrelizumab uygulamas�ndan ka��n�lmal�d�r.
Laktasyon d�nemi
Ocrelizumab�n ve metabolitlerinin insan s�t�ne kar���p kar��mad��� bilinmemektedir. Hayvan �al��malar�nda, ocrelizumab�n anne s�t�yle at�l�m� g�sterilmi�tir (bkz. B�l�m 5.3). Yenido�an ve bebeklerdeki risk g�z ard� edilemez. Kad�n hastalara tedavi s�ras�nda emzirmeyi kesmeleri �nerilmelidir.
�reme yetene�i/Fertilite
Sinomolgus maymunlar�nda erkek ve di�i do�urganl��� hakk�nda yap�lan �al��malara dayanan klinik �ncesi veriler, insanlara y�nelik �zel bir tehlikeye i�aret etmemektedir.
4.7. Ara� ve makine kullan�m� �zerindeki etkiler
OCREVUS'un ara� ve makine kullanma yetene�i �zerinde etkisi yoktur yada ihmal edilebilir d�zeydedir.
4.8. �stenmeyen etkiler
G�venlilik profilinin �zeti
En �nemli ve s�k bildirilen advers reaksiyonlar (A�R'ler); ��R'ler (RMS ve PPMS'te s�ras�yla %34,3 ve %40,1) , ve enfeksiyonlard�r (RMS ve PPMS'te s�ras�yla %58,5 ve %72,2) (bkz. B�l�m 4.4). .
Advers olaylar�n tablo halinde listesi
Klinik �al��malarda bildirilen ve spontan raporlamadan elde edilen advers reaksiyonlar a�a��da Tablo 2'de listelenmi�tir. Advers reaksiyonlar MedDRA sistem organ s�n�f� ve s�kl�k kategorilerine g�re listelenmi�tir. S�kl�k kategorileri bu �ekilde tan�mlanmaktad�r: �ok yayg�n (≥1/10), yayg�n (≥1/100 ila <1/10), yayg�n olmayan (≥1/1.000 ila <1/100), seyrek (≥1/10.000 ila <1/1.000), �ok seyrek (<1/10.000), ve bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor). Her bir s�kl�k grubunun i�inde istenmeyen etkiler azalan ciddiyet s�ras� ile sunulmaktad�r.
Tablo 2 OAdvers reaksiyonlar
A�R (MedDRA) Sistem Organ S�n�f� | �ok yayg�n | Yayg�n | Bilinmiyor |
Enfeksiyonlar ve enfestasyonlar |
�st solunum yolu enfeksiyonu, nazofarenjit, influenza | Sin�zit, bron�it, oral herpes, gastroenterit, solunum yolu enfeksiyonu, viral enfeksiyon, herpes zoster, konjunktivit, sel�lit |
|
Kan ve lenf sistemi hastal�klar� |
| N�tropeni | Ge� n�tropeni ba�lang�c� |
Solunum, g���s bozukluklar� ve mediastinal hastal�klar |
| �ks�r�k, nezle |
|
Ara�t�rmalar | Kan immunoglobulin M d����� | Kan immunoglobulin G d����� |
|
Yaralanma, zehirlenme ve prosed�rel komplikasyonlar |
�nf�zyon ile ili�kili 1 reaksiyonlar |
|
|

Se�ilen advers reaksiyonlar�n a��klamas�
�nf�zyonla ili�kili reaksiyonlar
RMS ve PPMS �al��malar�nda ��R'lerle ilgili semptomlar belirtilenlerle s�n�rl� olmamakla birlikte �unlar� i�erir: ka��nt�, d�k�nt�, �rtiker, eritem, y�zde k�zar�kl�k, hipotansiyon, ate�, yorgunluk, ba� a�r�s�, ba� d�nmesi, bo�azda tahri�, orofaringeal a�r�, dispne, faringeal veya laringeal �dem, bulant�, ta�ikardi. Kontroll� �al��malarda �l�mc�l ��R'ler olmam��t�r. Ek olarak, ��R'lerle ilgili semptomlara pazarlama sonras� verilerde anafilaksi dahil edilmi�tir.
Aktif kontroll� (RMS) klinik �al��malarda, interferon beta-1a tedavi grubunda (plasebo inf�zyonu) %9,9'luk insidansa k�yasla, ocrelizumab ile tedavi edilen grupta %34,3'l�k genel insidansla ��R'ler en yayg�n advers reaksiyondur. ��R'lerin insidans�, 1. doz, 1. inf�zyon (%27,5) s�ras�nda en y�ksek d�zeyde olmu� ve zamanla 4. dozda <%10'a d��m��t�r. Her iki tedavi grubunda ��R'lerin �o�unlu�u hafif ila orta �iddette olmu�tur. ocrelizumab ile tedavi edilen hastalar s�ras�yla %21,7 ve %10,1'i hafif veya orta, % 2,4'� �iddetli ��R ve % 0,1 hayat� tehdit eden ��R deneyimlemi�tir.
Plasebo kontroll� (PPMS) klinik �al��mada, plasebo grubundaki %25,5'lik insidansa k�yasla, ocrelizumab ile tedavi edilen grupta %40,1'lik genel insidansla ��R'ler en yayg�n advers reaksiyondur. ��R'lerin insidans�, 1. doz, 1. inf�zyon (%27,4) s�ras�nda en y�ksek d�zeyde olmu� ve zamanla 4. dozda <%10'a d��m��t�r. Her grupta her dozun ilk inf�zyonunda o dozun ikinci inf�zyonuna k�yasla daha b�y�k bir hasta oran� ��R'ler ya�am��t�r. ��R'lerin �o�unlu�u hafif ila orta �iddetli olmu�tur. Ocrelizumab ile tedavi edilen hastalar s�ras�yla %26,7 ve %11,9'u hafif veya orta �iddette, % 1,4'� �iddetli ��R deneyimlemi�tir. Hayat� tehdit eden ��R g�r�lmemi�tir (bkz. B�l�m 4.4).
Sonraki dozlar i�in alternatif h�zland�r�lm�� inf�zyon
Ataklarla seyreden Multipl Sklerozlu (RMS) hastalarda daha k�sa (2 saatlik) ocrelizumab inf�zyonlar�n�n g�venlilik profilini karakterize etmek i�in tasarlanm�� bir �al��mada (MA30143 H�zland�r�lm�� �nf�zyon �al��mas�), ��R'lerin g�r�lme s�kl���, yo�unlu�u ve tipleri 3,5 saat s�ren inf�zyon ile g�r�len ��R profili ile uyumlu bulunmu�tur (bkz B�l�m 5.1). Her iki inf�zyon grubunda da ihtiya� duyulan genel m�dahale say�s� d���kt�r, ancak 3,5 saatlik inf�zyon grubuna k�yasla, k�sa (2 saatlik) inf�zyon grubunda ��R'leri y�netebilmek i�in daha fazla m�dahale (yava�lama veya ge�ici kesintiler) gerekmi�tir (s�ras�yla% 8,7'ye kar��% 4,8).
Enfeksiyon
Aktif kontroll� RMS �al��malar�nda, ocrelizumab alan hastalar�n %58,5'inde ve interferon beta- 1a alan hastalar�n %52,5'inde enfeksiyon g�zlenmi�tir. ocrelizumab alan hastalar�n %1,3'�ne kar��l�k interferon beta-1a alan hastalar�n %2,9'unda ciddi enfeksiyon g�zlenmi�tir. Plasebo kontroll� PPMS �al��mas�nda ocrelizumab alan hastalar�n %72,2'sinde ve plasebo alan hastalar�n %69,9'unda enfeksiyon g�zlenmi�tir. Ocrelizumab alan hastalar�n %6,2'sine kar��l�k plasebo alan hastalar�n %6,7'sinde ciddi enfeksiyon meydana gelmi�tir. Hem RMS hem de PPMS �al��malar�nda t�m hastalar a��k etiketli fazda ocrelizumaba ge�mi�tir. RMS'de ciddi enfeksiyon riskinde art�� 2. ve 3. y�llar aras�nda g�zlenmi� olup takip eden y�llarda g�zlenmemi�tir. PPMS'de ise herhangi bir art�� g�zlenmemi�tir.
Solunum yolu enfeksiyonlar�
Solunum yolu enfeksiyonlar�n�n oran�, interferon beta-1a ve plaseboya k�yasla ocrelizumab ile tedavi edilen hastalarda daha y�ksek olmu�tur.
RMS klinik �al��malar�nda ocrelizumab ile tedavi edilen hastalar�n %39,9'u ve interferon beta-1a ile tedavi edilen hastalar�n % 33,2 '� �st solunum yollar� enfeksiyonu; ocrelizumab ile tedavi edilen hastalar�n %7,5'i ve interferon beta-1a ile tedavi edilen hastalar�n %5,2'si alt solunum yollar� enfeksiyonu deneyimlemi�tir. PPMS klinik �al��mas�nda, ocrelizumab ile tedavi edilen hastalar�n %48,8'i ve plasebo alan hastalar�n %42,7'si �st solunum yollar� enfeksiyonu; ocrelizumab ile tedavi edilen hastalar�n %9,9'u ve plasebo alan hastalar�n %9,2'si alt solunum yollar� enfeksiyonu deneyimlemi�tir. Ocrelizumab ile tedavi edilen hastalar�n solunum yollar� enfeksiyonu a��rl�kl� olarak hafif ila orta olarak raporlanm��t�r (%80-90).
Herpes
Aktif kontroll� (RMS) klinik �al��malarda, herpes enfeksiyonlar�, ocrelizumab ile tedavi edilen hastalarda interferon beta-1a ile tedavi edilen hastalara k�yasla daha s�k bildirilmi�tir: herpes zoster (%2,1 ile %1), herpes simpleks (% 0,7 ile % 0,1) ve oral herpes (%3 ile %2,2), genital herpes (%0,1 ile %0) ve herpes vir�s enfeksiyonu (%0,1 ile %0). T�m enfeksiyonlar, bir tane Derece 3 hari�, hafif ila orta �iddetli olmu� ve hastalar standart tedavi uygulanmas�yla iyile�mi�lerdir.
Plasebo kontroll� (PPMS) klinik �al��mada, oral herpesi olan hastalar ocrelizumab tedavi kolunda, plaseboya g�re daha s�k rapor edilmi�tir (%2,7'ye kar�� %0,8).
Laboratuvar anormallikleri �mmunoglobulinler
Ocrelizumab tedavisi, a��rl�kl� olarak IgM'deki azalmaya ba�l� olarak �al��malar�n kontroll� d�neminde toplam immunoglobulinlerde azalmayla sonu�lanm��t�r. Klinik �al��ma verileri, IgG'de s�rekli azalma (ve IgM ve IgA i�in daha az) ile ciddi enfeksiyonlar aras�nda bir ili�ki
oldu�unu g�stermi�tir.
Lenfositler
RMS'de, interferon beta-1a ile tedavi edilen hastalar�n %32,6's�na kar��l�k ocrelizumab ile tedavi edilen hastalar�n %20,7'sinde lenfositin NAS'�n (normalin alt s�n�r�) alt�na d��t��� g�zlenmi�tir. PPMS'de plaseboyla tedavi edilen hastalar�n %11,7'sine kar��l�k ocrelizumab ile tedavi edilen hastalar�n %26,3'�nde lenfositlerin NAS'�n alt�na d��t��� g�zlenmi�tir.
Ocrelizumab ile tedavi edilen hastalarda bildirilen bu d����lerin �o�unun �iddet olarak Derece
1 (<NSA– 800 h�cre/mm) ve Derece 2 (500-800 h�cre/mm) oldu�u g�r�lm��t�r. Ocrelizumab grubundaki hastalar�n yakla��k %1'inde Derece 3 lenfopeni (200-500 h�cre/mm) oldu�u g�r�lm��t�r. Hastalar�n hi�birinde Derece 4 lenfopeni (<200 h�cre/mm) bildirilmemi�tir.
Ocrelizumabla tedavi edilen hastalarda do�rulanm�� total lenfosit say�m� d���� epizotlar� esnas�nda ciddi enfeksiyonlar�n oran�nda art�� g�zlenmi�tir. Ciddi enfeksiyon say�s�n�n, kesin sonu�lara varmak i�in �ok d���k oldu�u g�r�lm��t�r.
N�trofil
Aktif kontroll� (RMS) tedavi d�neminde, interferon beta-1a ile tedavi edilen hastalar�n %40,9'una k�yasla, ocrelizumab ile tedavi edilen hastalar�n %14,7'sinde n�trofillerin NSA'�n alt�na d��t��� g�zlenmi�tir. Plasebo kontroll� (PPMS) klinik �al��mada, n�trofil seviyelerinde azalma g�r�len ocrelizumab hastalar�n�n oran�, plasebo hastalar�na (%12,9) k�yasla daha y�ksek (%10,0) olmu�tur. Plasebo grubundaki hastalar�n %1,3'�ne kar��l�k ocrelizumab grubunda daha y�ksek oranda hastada (%4,3) yada ≥Derece 2 n�tropeni g�r�lm��; plasebo grubundaki hastalar�n %0'�na kar��l�k ocrelizumab grubundaki hastalar�n yakla��k %1'inde Derece 4 n�tropeni g�r�lm��t�r.
N�trofil d����lerinin �o�unun ge�ici nitelikte (sadece ocrelizumab ile tedavi edilen belirli bir hastada bir defa g�zlenmi�tir) ve �iddet olarak Derece 1 ve 2 (s�ras�yla <NAS ve 1500 h�cre/mm aras�nda ve 1000-1500 h�cre/mm) oldu�u belirlenmi�tir. Genel olarak, ocrelizumab grubundaki hastalar�n yakla��k %1'inde Derece 3 veya 4 n�tropeni vard�. Derece 3 (500 – 1000 h�cre/mm) n�tropenili bir hasta ve Derece 4(<500 h�cre/mm) n�tropenili bir hasta gran�losit-koloni uyar�c� fakt�rle spesifik tedaviye ihtiya� duymu� ve epizot sonras�nda ocrelizumab tedavisine devam etmi�tir. N�tropeni, ocrelizumab uygulamas�ndan birka� ay sonra ortaya ��kabilir (bkz. B�l�m 4.4).
Di�er
2000 mg ocrelizumab alan ba�ka bir hasta ise en son inf�zyondan 12 hafta sonra uygulanan manyetik rezonans g�r�nt�lemesinin (MRG) ard�ndan etiyolojisi bilinmeyen sistemik enflamatuvar yan�t sendromu (SEYS) nedeniyle �lm��t�r; MRG i�in uygulanan gadolinyumlu kontrast maddeye kar�� geli�en anafilaktoid reaksiyon SEYS'nin ortaya ��kmas�na katk�da bulunmu� olabilir.
��pheli advers reaksiyonlar�n raporlanmas�
Ruhsatland�rma sonras� ��pheli ila� advers reaksiyonlar�n�n raporlanmas� b�y�k �nem ta��maktad�r. Raporlama yap�lmas�, ilac�n yarar/risk dengesinin s�rekli olarak izlenmesine olanak sa�lar. Sa�l�k mesle�i mensuplar�n�n herhangi bir ��pheli advers reaksiyonu T�rkiye Farmakovijilans Merkezi'ne (T�FAM) bildirmeleri gerekmektedir (www.titck.gov.tr: e-posta: tufam@titck.gov.tr, tel: 0 800 314 00 08; faks: 0 312 218 35 99).
4.9. Doz a��m� ve tedavisi
Onayl� intraven�z ocrelizumab dozundan daha y�ksek dozlarda klinik �al��ma deneyimi s�n�rl�d�r. MS hastalar�nda bug�ne kadar test edilen en y�ksek doz, 2 hafta arayla iki 1000 mg intraven�z inf�zyon olarak uygulanan 2000 mg'd�r (RRMS'de Faz II doz bulma �al��mas�). Advers reaksiyonlar�, merkezi klinik �al��malarda g�venlilik profiliyle tutarl� olmu�tur.
Doz a��m� olmas� durumunda spesifik bir antidot yoktur; inf�zyon hemen durdurulmal� ve hasta ��R'ler a��s�ndan g�zlenmelidir (bkz. B�l�m 4.4).
5. FARMAKOLOJ�K �ZELL�KLER
5.1. Farmakodinamik �zellikler
Farmakoterap�tik grup: Antineoplastik ve �mm�nomod�lat�r Ajanlar, �mm�nosupresanlar, Selektif �mm�nosupresanlar
ATC kodu: L04AA36
Etki mekanizmas�
Ocrelizumab, CD20 eksprese eden B h�crelerini se�ici olarak hedefleyen bir rekombinant h�manize monoklonal antikordur.
CD20, �n-B h�creleri, olgun ve bellek B h�crelerinde bulunan ama lenfoid k�k h�creleri ve plazma h�crelerinde eksprese edilmeyen bir h�cre y�zeyi antijenidir.
Ocrelizumab�n MS'te terap�tik klinik etkilerini g�sterdi�i kesin mekanizmalar tam olarak a��klanmam��t�r ama CD20 eksprese eden B h�crelerinin say�s� ve fonksiyonunda azalmayla immunomod�lasyona neden oldu�u varsay�l�r. H�cre y�zeyindeki ba�lanman�n ard�ndan, ocrelizumab, antikor ba��ml� sell�ler fagositoz (ADCP), antikor ba��ml� sell�ler sitotoksisite (ADCC), kompleman ba��ml� sitotoksisite (CDC) ve apoptoz arac�l���yla CD20 eksprese eden B h�crelerini se�ici olarak t�ketir. B h�cresi rekonstit�syon kapasitesi ve �nceden var olan humoral ba����kl�k korunur. Ayr�ca do�u�tan ba����kl�k ve toplam T h�cresi say�lar� etkilenmez.
Farmakodinamik etkiler
Ocrelizumab ile tedavi, beklenen bir farmakolojik etki olarak, tedaviden 14 g�n sonra (de�erlendirmenin ilk zaman noktas�) kanda CD19+ B h�crelerinin h�zl� t�kenmesine yol a�ar. Bu, tedavi d�nemi boyunca s�rd�r�lm��t�r. Ocrelizumab�n varl��� miktar tayiniyle CD20'nin tan�nmas�n� engelledi�i i�in, B h�cre say�mlar� i�in CD19 kullan�l�r.
Faz III �al��malarda, her ocrelizumab dozunun aras�nda, hastalar�n %5'ine kadar� en az bir zaman noktas�nda B h�cresi deplesyonu (> normalin alt s�n�r� (NAS) veya ba�lang��) sergilemi�tir. B h�cresi t�kenmesinin derecesi ve s�resi, PPMS ve RMS �al��malar�nda tutarl� olmu�tur.
Son Ocrelizumab inf�zyonundan sonraki en uzun takip s�resi (Faz II WA21493, N=51), B h�cresi �o�almas�na kadar ge�en medyan s�renin (hangisinin daha �nce oldu�una ba�l� olarak ba�lang�ca veya LLN'ye geri d�nm��t�r) 72 hafta (27 - 175 haftal�k aral�k) oldu�unu g�stermektedir. T�m hastalar�n y�zde doksan�nda, son inf�zyondan yakla��k iki bu�uk y�l sonra B h�creleri LLN veya ba�lang�ca g�re �o�alm��t�r.
Klinik etkililik ve g�venlilik
Ataklarla seyreden multipl skleroz formlar� (RMS)
Ocrelizumab�n etkilili�i ve g�venlili�i, ataklarla seyreden MS formlar� olan hastalarda tasar�mlar� ayn� olan iki randomize, �ift k�r, �ift plasebolu, aktif komparat�r kontroll� klinik �al��mada (WA21092 ve WA21093) de�erlendirilmi�tir (2010 McDonald kriterlerine uygun olarak). �al��ma tasar�m� ve �al��ma pop�lasyonunun ba�lang�� �zellikleri Tablo 3'te �zetlenmektedir.
Demografi ve ba�lang�� �zellikleri a��s�ndan her iki tedavi grubu iyi dengelenmi�tir. Ocrelizumab alan hastalara (Grup A) 6 ayda bir 600 mg (1. Doz 2 hafta arayla 2 x 300 mg intraven�z inf�zyonlar halinde) verilmi� ve sonraki dozlar tek bir 600 mg intraven�z inf�zyon olarak uygulanm��t�r. Grup B'deki hastalara haftada 3 kez subk�tan (S.C.) enjeksiyonla �nterferon beta-1a 44 mcg verilmi�tir.
Tablo 3 �al��ma Tasar�m� ve Demografik �zellikler
| �al��ma 1 | �al��ma 2 | ||
�al��ma ad� | WA21092 (OPERA I) (n=821) | WA21093 (OPERA II) | ||
�al��ma tasar�m� | ||||
�al��ma pop�lasyonu | Ataklarla seyreden MS formlar� g�r�len hastalar | |||
Taramadaki hastal�k �yk�s� | �ki y�l i�inde en az iki relaps veya bir y�l i�inde bir relaps; EDSS 0 ve 5,5 (5,5 dahil) aras�nda, | |||
�al��ma s�resi | 2 y�l | |||
Tedavi gruplar� | Grup A: 600 mg ocrelizumab Grup B: 44 mcg SC interferon beta-1a (IFN) | |||
Ba�lang��taki �zellikler | Ocrelizumab 600 mg (n=410) | IFN 44 mcg (n=411) | Ocrelizumab 600 mg (n=417) | IFN 44 mcg (n=418) |
Medyan ya� (y�l) | 37,1 | 36,9 | 37,2 | 37,4 |
Dahil edilen ya� aral��� (y�l) | 18-56 | 18-55 | 18-55 | 18-55 |
Cinsiyet da��l�m� (%erkek/%kad�n) | 34,1/65,9 | 33,8/66,2 | 35/65 | 33/67 |
Tan�dan bu yana ge�en ortalama/medyan hastal�k s�resi (y�l) | 3,82/1,53 | 3,71/1,.57 | 4,15/2,1 | 4,13/1,84 |
Daha �nce DMT almam�� hastalar�n %'si** | 73,4 | 71 | 72,7 | 74,9 |
Ge�en y�ldaki ortalama relaps say�s� | 1,31 | 1,33 | 1,32 | 1,34 |
T1 lezyonunda Gd tutulumlu hastalar�n oran� | 42,5 | 38,1 | 39,0 | 41,4 |
Ortalama EDSS* | 2,82 | 2,71 | 2,73 | 2,79 |
* Geni�letilmi� Yeti Yitimi Durumu �l�e�i
**Randomizasyondan �nceki 2 sene i�inde bir hastal�k modifiye edici tedavi (DMT) ile tedavi edilmemi� hastalar
Temel klinik ve MRG etkilili�i sonu�lar� Tablo 4 ve �ekil 1'de sunulmaktad�r.
Bu �al��malar�n sonu�lar�, interferon beta-1a 44mcg SC'ye k�yasla ocrelizumab�n relapslar�, MRG ile �l��len klinik ve subklinik hastal�k aktivitesi ve hastal�k progresyonunu �nemli �l��de bask�lad���n� g�stermektedir.
Tablo 4 WA21092 ve WA21093 �al��malar�ndan Temel Klinik ve MRG Sonlan�m Noktalar� (RMS)
Sonlan�m Noktalar� | �al��ma 1: WA21092 (OPERA I) | �al��ma 2: WA21093 | |||
Ocrelizumab 600 mg (n=410) | IFN 44 mcg (n=411) | Ocrelizumab 600 mg (n=417) | IFN 44 mcg (n=418) | ||
Klinik Sonlan�m Noktalar� |
| ||||
Y�ll�k Relaps Oran� (primer sonlan�m noktas�) R�latif Azalma | 0-156 | 0,292 | 0,155 | 0,290 | |
%46 (p<0,0001) | %47 (p<0,0001) | ||||
12 Haftal�k Do�rulanm�� Yeti Yitimi Progresyonu G�r�len Hasta Oran� Riskte Azalma (Birle�tirilmi� Analiz) Riskte Azalma (Ayr� Ayr� �al��malar) | %9,8 Ocrelizumab - %15,2 IFN %40 (p=0,0006) | ||||
%43 (p=0,0139) | %37 (p=0,0169) | ||||
24 Haftal�k Do�rulanm�� Yeti Yitimi Progresyonu G�r�len Hasta Oran� (CDP) Riskte Azalma (Birle�tirilmi� Analiz) Riskte Azalma (Ayr� Ayr� �al��malar) | %7,6 Ocrelizumab - %12,0 IFN %40 (p=0,0025) | ||||
%43 (p=0,0278) | %37 (p=0,0370) | ||||
En az 12 haftal�k Do�rulanm�� Yeti Yitimi �yile�mesi g�r�len hasta oran�(Birle�tirilmi�) R�latif Art�� (Birle�tirilmi� Analiz) R�latif Art�� (Ayr� Ayr� �al��malar) | %20,7 Ocrelizumab - %15,6 IFN | ||||
%33 (p=0,0194) | |||||
%61 (p=0,0106) | %14 (p=0,4019) | ||||
96 haftada relaps g�zlenmeyen hastalar�n oran� | %80,4 | %66,7 | %78,9 | %64,3 | |
(p=0,0001) | (p<0,0001) | ||||
Hastal�k Aktivitesi Kan�t� G�r�lmeyen (NEDA) Hasta Oran� R�latif Art�� | %48 | %29 | %48 | %25 | |
%64 (p<0,0001) | %89 (p<0,0001) | ||||
MRG Sonlan�m Noktalar� |
| ||||
MRG taramas� ba��na ortalama Gd tutulumlu T1 lezyon say�s� R�latif Azalma | 0,016 | 0,286 | 0,021 | 0,416 | |
%94 (p<0,0001) | %95 (p<0,0001) | ||||
MRG taramas� ba��na yeni ve/veya b�y�yen T2 hiperintens lezyonlar�n ortalama say�s� R�latif Azalma | 0,323 | 1,413 | 0,325 | 1,904 | |
%77 (p<0,0001) | %83 (p<0,0001) | ||||
|
|
|
|
| |
|
| ||||
24. hafta ila 96. hafta aras�nda beyin hacmindeki de�i�iklik oran� Beyin hacmi kayb�nda r�latif azalma | -0,572 | -0,741 | -0,638 | -0,750 | |
%22,8 (p=0,0042) | %14,9 (p=0,0900) | ||||
tahminleri
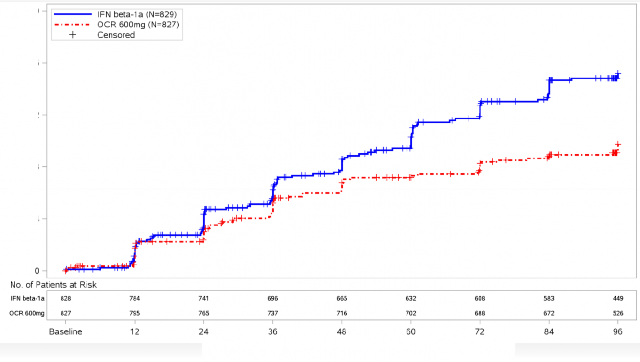
�ekil 1: En Az 12 Hafta Boyunca S�rd�r�len Do�rulanm�� Yeti Yitimi Progresyonunun Ba�lang�c�na Kadar Ge�en S�re ve �ift-K�r Tedavi D�nemi Boyunca Meydana Gelen �lk N�rolojik K�t�le�me Olay�na �li�kin Kaplan-Meier Grafi�i* (Birle�tirilmi� ITT pop�lasyonu)*
*OPERA I ve II'nin �nceden belirlenmi� birle�tirilmi� analizi
En az 12 hafta s�rd�r�len CDP'ye kadar ge�en s�renin �nceden belirlenen havuzlanm�� analizlerinin sonu�lar� (interferon beta-1a'ya k�yasla ocrelizumab i�in %40 risk azalt�m�, (p=0,0006), en az 24 hafta s�rd�r�len sonu�larla (interferon beta-1a'ya k�yasla ocrelizumab i�in %40 risk azalt�m�, p=0,0025) son derece tutarl� olmu�tur.
�al��malarda aktif hastal��� olan hastalar dahil edilmi�tir. Bu hastalarda, klinik ve radyolojik a��dan aktif, tedavi naif ve �nceki tedavilerine yetersiz cevap geli�tirmi� hastalar dahildir. �ok aktif ve aktif hastal�k durumu da dahil olmak �zere farkl� ba�lang�� seviyelerinde hastal�k aktiviteleri olan hasta pop�lasyonu analizleri, ocrelizumab etkilili�inin ARR'de ve CDP ile 12 haftada genel pop�lasyon ile ayn� oldu�unu ortaya koymu�tur.
Primer progresif multipl skleroz (PPMS)
Ocrelizumab�n etkililik ve g�venlili�i ayr�ca ana dahil etme kriterlerine (18-55 ya� (bu ya�lar dahil), taramada 3– 6,5 puan aras� EDSS, MS semptomlar�n�n ortaya ��k���ndan itibaren ge�en hastal�k s�resi: taramadaki EDSS puan� ≤5olan hastalarda <10 y�l ve EDSS puan� <5olan hastalarda <15 y�l) g�re hastal���n erken evresinde olan primer progresif MS'li hastalara y�nelik randomize, �ift-k�r, plasebo-kontroll� bir �al��mada (�al��ma WA25046) de�erlendirilmi�tir. Hastal�k aktivitesi bak�m�ndan, progresif MS'te dahi enflamatuvar etkinli�e �zg� �zellikler g�r�nt�lemeyle ili�kili olabilir (T1 Gd-kontrast tutan lezyonlar ve/veya aktif (yeni veya b�y�yen) T2 lezyonlar�). B�t�n hastalarda enflamatuvar etkinli�i do�rulamak i�in MRG bulgular� kullan�lmal�d�r. 55 ya��n �zerindeki hastalar incelenmemi�tir. �al��ma tasar�m� ve �al��ma pop�lasyonunun ba�lang�� �zellikleri Tablo 5'te sunulmu�tur.
Demografik �zellikler ve ba�lang�� �zelliklerinin iki tedavi grubu aras�nda dengeli oldu�u g�r�lm��t�r. Kraniyal MRG T1 Gd-kontrast tutan lezyonlar veya T2 lezyonlar� olarak enflamatuvar etkinli�e �zg� g�r�nt�leme �zellikleri g�stermi�tir.
Faz III PPMS �al��mas� s�ras�nda, hastalar her 6 ayda bir 600 mg ocrelizumab dozunu, iki hafta ara ile uygulanan 2 adet 300 mg'l�k inf�zyonlar �eklinde alm��t�r. RMS'de 600 mg inf�zyonlar ve PPMS'de 300 mg x 2 inf�zyonlar uyumluPK/PD profilleri g�stermi�tir. �nf�zyon ba��na ��R profilleri tek seferde 600 mg veya iki hafta arayla 300 mg doz uygulanmas�ndan ba��ms�z olarak benzerdir (bkz. B�l�m 4.8 ve 5.2). Ancak 2x300 mg doz rejimi ile toplamdaki inf�zyon say�s� daha y�ksek oldu�unda, toplam ��R say�s� daha y�ksektir. Bu nedenle, Doz 1'den sonra toplam inf�zyon say�s�n� d���rmek i�in (metilprednisolon ve antihistaminik ile e� zamanl� uygulanmas� s�ras�nda) ocrelizumab�n 600 mg'l�k tek bir inf�zyon olarak uygulanmas� tavsiye edilir (bkz. B�l�m 4.2).
Tablo 5 WA25046 �al��mas�n�n �al��ma tasar�m�, demografik ve ba�lang�� �zellikleri
�al��ma ad� | WA25046 �al��mas� ORATORIO (n=732) | |
�al��ma tasar�m� | ||
�al��ma pop�lasyonu | Primer progresif MS formu g�r�len hastalar | |
�al��ma s�resi | Olaya dayal� (Minimum 120 hafta ve 253 do�rulanm�� yeti yitimi progresyonu olay�) (Medyan takip s�resi: Ocrelizumab 3 y�l, Plasebo 2,8 y�l | |
Taramadaki hastal�k �yk�s� | Ya� 18-55, EDSS 3 ila 6,5 | |
Tedavi gruplar� | Grup A: 600 mg Ocrelizumab Grup B: Plasebo, 2:1 randomizasyon | |
Ba�lang��taki �zellikler | 600 mg Ocrelizumab (n=488) | Plasebo (n=244) |
Ortalama Ya� (y�l) | 44,7 | 44,4 |
Dahil edilen ya� aral��� | 20-56 | 18-56 |
Cinsiyet da��l�m� (%erkek/%kad�n) | 51,4/48,6 | 49,2/50,8 |
PPMS tan�s�ndan bu yana ge�en ortalama/medyan hastal�k s�resi (y�l) | 2,9/1,6 | 2,8/1,3 |
Ortalama EDSS | 4,7 | 4,7 |
Temel klinik ve MRG etkilili�i sonu�lar� Tablo 6 ve �ekil 2'de sunulmaktad�r.
Bu �al��man�n sonu�lar�, ocrelizumab�n plaseboya k�yasla hastal�k progresyonunu �nemli �l��de geciktirdi�ini ve y�r�me h�z�ndaki k�t�le�meyi azaltt���n� g�stermektedir.
Tablo 6 WA25046 �al��mas�ndan (PPMS) Temel Klinik ve MRG Sonlan�m Noktalar�
| �al��ma 3 | |
Sonlan�m Noktalar� | WA25046 (ORATORIO) | |
Ocrelizumab 600 mg (n=488) | Plasebo (n=244) | |
Klinik Sonlan�m Noktalar� | ||
Primer etkililik sonlan�m noktas� 12 Haftal�k Do�rulanm�� Yeti Yitimi Progresyonu G�r�len Hasta Oran� (primer sonlan�m noktas�) Riskte azalma | %30,2 | %34 |
%24 (p=0,0321) | ||
24 Haftal�k Do�rulanm�� Yeti Yitimi Progresyonu G�r�len Hasta Oran� Riskte azalma | %28,3 | %32,7 |
%25 (p=0,0365) | ||
Ba�lang�ca k�yasla 120. Haftada S�reli 25 Ad�m Y�r�me Testindeki De�i�iklik Oran� Y�r�me s�resindeki progresyon oran�nda r�latif azalma | 38,9 | 55,1 |
%29,4 (p=0,0404) | ||
MRG Sonlan�m Noktalar� | ||
Ba�lang�ca k�yasla 120. Haftada T2 hiperintens lezyon hacmindeki de�i�iklik oran� | -3,4 | 7,4 |
(p< 0,0001) | ||
24. hafta ila 120. hafta aras�nda beyin hacmindeki de�i�iklik oran� Beyin hacmi kay�p oran�nda r�latif azalma | -0,902 | -1,093 |
%17,5 (p=0,0206) | ||
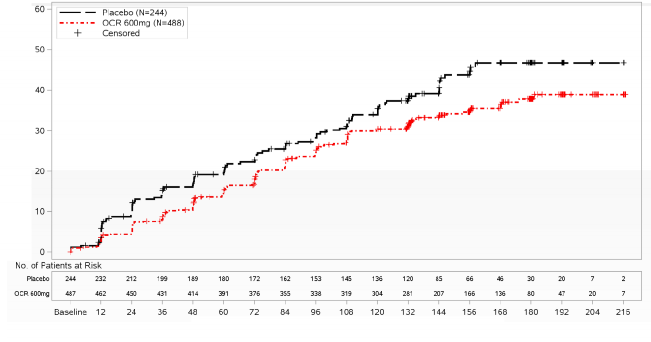
CDP riskinde %24 azalma HR (%95 GA): 0,76 (0,59,
0,98); p=0,0321
�ekil 2: �ift K�r Tedavi D�neminde Meydana Gelen �lk N�rolojik K�t�le�me Olay�yla En Az 12 Hafta S�rd�r�len Do�rulanm�� Sakatl�k Progresyonunun Ba�lang�c�na Kadar Ge�en S�renin Kaplan-Meier Grafi�i (ITT Pop�lasyonu)*
* Bu analizdeki t�m hastalar en az 120 hafta boyunca takip edilmi�tir. Birincil analiz de�erlendirilen t�m olaylara dayanmaktad�r.
Birincil sonlan�m noktas�na y�nelik �nceden tan�mlanm��, yeterli g�ce sahip olmayan alt grup analizi, gen� veya ba�lang��ta T1 Gd-kontrast tutan lezyonlar� olan hastalar�n ya�l� veya T1 Gd-kontrast tutan lezyonlar� olmayan hastalara k�yasla tedaviden daha fazla fayda sa�lad���n� g�stermektedir (≤ 45 ya�: HR 0,64 [0,45, 0,92], >45 ya�: HR 0,88 [0,62, 1,26]; ba�lang��ta T1 Gd-kontrast tutan lezyonlar� olan hastalar: HR 0,65 [0,4-1,06], T1 Gd-kontrast tutan lezyonlar� olmayan hastalar: HR 0,84 [0,62-1,13]).
Ayr�ca, post-hoc analizler ba�lang��ta T1 Gd-kontrast tutan lezyonlar� olan gen� hastalarda tedavinin daha iyi bir etki g�sterdi�ini ortaya koymu�tur (≤ 45 ya�: HR 0,52 [0,27-1]; ≤ 46 ya� [WA25046 �al��mas�ndaki medyan ya�]; HR 0,48 [0,25-0,92]; <51 ya�: HR 0,53 [0,31-0,89]).
A��k Etiket Uzant�s�na (AEU) devam etmeden �nce veya �al��ma tedavisinden ayr�lana kadar ek olarak yakla��k 9 ayl�k kontroll� takip ve �ift k�r tedavi i�eren Geni�letilmi� Kontroll� D�nemde (GKD) post-hoc analizler ger�ekle�tirilmi�tir. 24 haftal�k Do�rulanm�� Engellilik Progresyonunun (CDP) EDSS≥7 olan hastalar�n oran� (EDSS≥7'nin 24W-CDP'si, tekerlekli sandalyeye kadar ge�en s�re), tedavinin 144. haftas�nda plasebo grubunda %9,1 iken, ocrelizumab grubunda %4,8 olmu� ve bu oran GKD s�ras�nda tekerlekli sandalyeye ge�i� s�resinin %47 risk azalmas�na (HR 0,53, [0,31, 0,92]) neden olmu�tur. Bu sonu�lar do�alar� gere�i ke�if ama�l� oldu�undan ve k�rleme kald�r�ld�ktan sonra veriler i�erdi�inden, dikkatle yorumlanmal�d�r.
H�zland�r�lm�� inf�zyon �al��mas�
H�zland�r�lm�� (2 saatlik) ocrelizumab inf�zyonunun g�venlili�i, di�er hastal�k modifiye edici tedavilere naif olan Ataklarla Seyreden Multipl Sklerozlu (RMS) hastalarda MA30143 (Ensemble) �al��mas�n�n alt kolu olan prospektif, �ok merkezli, randomize, �ift k�r, kontroll�,
paralel kollu bir �al��ma ile de�erlendirilmi�tir. �lk doz 14 g�n arayla iki 300 mg inf�zyon (toplam 600 mg) olarak uygulanm��t�r. Hastalar ikinci dozlar�ndan sonra (Doz 2 ila 6) 1: 1 oran�nda ya ocrelizumab�n 24 haftada bir uygulanan yakla��k 3.5 saat boyunca inf�ze edilmi� konvansiyonel inf�zyon grubuna ya da ocrelizumab�n 24 haftada bir uygulanan yakla��k 2 saat s�ren daha k�sa inf�zyon s�resi grubuna randomize edilmi�tir. Randomizasyon, b�lgeye ve hastalar�n ilk randomize edildi�i doza g�re tabakaland�r�lm��t�r.
Birincil sonlan�m noktas�, ilk randomize inf�zyonu s�ras�nda yada takip eden 24 saat i�inde ��R meydana gelen hastalar�n oran�d�r. Primer analiz 580 hasta randomize edildi�inde yap�lm��t�r. �lk randomize inf�zyon s�ras�nda yada takiben 24 saat i�inde ��R meydana gelen hastalar�n oran�, h�zland�r�lm�� inf�zyonda % 24,6 iken, konvansiyonel inf�zyon grubunda % 23,1 olmu�tur. Tabakal� grup fark� benzerdir. Genel olarak, t�m randomize dozlarda, ��R'lerin �o�unlu�u hafif veya orta d�zeydeydi ve her iki grupta birer ��R olmak �zere sadece iki ��R �iddetli olmu�tur. Hayat� tehdit eden, �l�mc�l veya ciddi ��R'lerg�r�lmemi�tir.
�mmunojenisite
MS �al��malar�ndaki (WA21092, WA21093 ve WA25046) hastalar bir�ok zaman noktas�nda (ba�lang�� ve �al��ma s�resi boyunca tedavi sonras� 6 ayda bir) anti-ila� antikorlar� (A�A'lar) a��s�ndan test edilmi�tir. Ocrelizumab ile tedavi edilen 1311 hastadan 12'si (~%1) tedaviyle ortaya ��kan A�A'lar a��s�ndan pozitif sonu� vermi�, bunlar�n 2'si n�tralize edici antikorlar a��s�ndan pozitif sonu� vermi�tir. Ocrelizumab ile ili�kili d���k A�A insidans� d���n�l�rse, tedaviyle ortaya ��kan A�A'lar�n g�venlilik ve etkililik �zerindeki etkisi de�erlendirilemez.
�mmunizasyonlar
RMS hastalar�nda ger�ekle�tirilen a��k etiketli, randomize bir �al��mada (N=102), a�� uygulamas�ndan sonra 8. haftada tetanoz a��s�na pozitif yan�t veren hastalar�n oran�, kontrol grubunda %54,5'e k�yasla ocrelizumab grubunda %23,9 olarak bulunmu�tur (interferon-beta haricinde hastal��� modifiye edici herhangi bir tedavi mevcut de�ildir). 8. haftada anti-tetanoz toksoidine spesifik antikor titresi geometrik ortalamas� s�ras�yla 3,74 ve 9,81 IU/mL olarak tespit edilmi�tir. A�� uygulamas�ndan sonra 4. haftada 23-PPV'de bulunan ≥5 serotipe verilen pozitif yan�t oran�, kontrol grubunda %100 ve ocrelizumab grubunda %71,6'd�r. Ocrelizumab ile tedavi edilen hastalarda 23-PPV'den 4 hafta sonra uygulanan rapel a��s� (peki�tirme dozu/13-PCV), 23-PPV ile ortak 12 serotipe verilen yan�t� belirgin bir �ekilde artt�rmam��t�r. Be� influenza su�una kar�� seroprotektif titreleri bulunan hastalar�n y�zdesi, ocrelizumab ile tedavi edilen hastalarda ve kontrol grubunda s�ras�yla, a��lama �ncesinde %20,0-60,0 ve %16,7-43,8 ve a��lamadan 4 hafta sonra %55,6-80,0 ve %75,0-97,0 aral�klar�nda
bulunmu�tur. (bkz. B�l�m 4.4 ve 4.5)
5.2. Farmakokinetik �zellikler
Genel �zellikler
Emilim:
Ocrelizumab intraven�z inf�zyon olarak uygulan�r. Di�er uygulama yollar�yla �al��ma ger�ekle�tirilmemi�tir.
Da��l�m:
Merkezi da��t�m hacminin pop�lasyon farmakokineti�i tahmini 2,78 L olmu�tur. Periferal hacim ve kompartmanlar aras� klerens 2,68 L ve 0,294 L/g�n olarak tahmin edilmi�tir.
Biyotransformasyon:
Antikorlar a��rl�kl� olarak katabolizmayla (�r: peptid ve amino asitlere y�k�lma)
uzakla�t�r�ld���ndan, ocrelizumab�n metabolizmas� �zerinde do�rudan �al���lmam��t�r. Eliminasyon:
Sabit klerens 0,17 L/g�n tahmin edilirken, ba�lang��taki zamana ba��ml� klerens 0,0489 L/g�n olarak tahmin edilmi� ve 33 haftal�k yar� �m�rle birlikte d��m��t�r. Ocrelizumab�n terminal eliminasyon yar� �mr� 26 g�n olmu�tur.
Do�rusall�k/do�rusal olmayan durum:
MS �al��malar�nda ocrelizumab�n farmakokineti�i, zamana ba�l� klerens ve bir IgG1 monoklonal antikor i�in tipik PK parametreleri sergileyen, iki b�l�ml� bir modelle a��klanm��t�r.
Genel maruziyet (24 haftal�k dozlama aral�klar�nda EAA), ayn� dozun uyguland��� d���n�l�rse beklenece�i �zere, PPMS �al��malar�nda 2 x 300 mg ve RMS �al��malar�nda 1 x 600 mg'da ayn� olmu�tur. 600 mg ocrelizumab�n 4. dozundan sonra e�ri alt� alan (EAA) 3510 mcg/mL•g�n ve ortalama maksimum konsantrasyon (C) RMS'de (600 mg inf�zyon) 212 mcg/mL, PPMS'de (300 mg inf�zyonlar) 141 mcg/mL olmu�tur.
�zel pop�lasyonlara ili�kin ek bilgiler:
Pediyatrik pop�lasyon:
18 ya��n�n alt�ndaki �ocuklarda ocrelizumab�n farmakokineti�ini ara�t�rmak amac�yla �al��ma y�r�t�lmemi�tir.
Geriyatrik pop�lasyon:
55 ya� ve �zerindeki hastalarda ocrelizumab�n farmakokineti�ini ara�t�rmak amac�yla �al��ma y�r�t�lmemi�tir.
B�brek yetmezli�i :
Resmi farmakokinetik �al��ma y�r�t�lmemi�tir. Hafif b�brek yetmezli�i olan hastalar klinik �al��malara dahil edilmi� ve bu hastalarda ocrelizumab�n farmakokineti�inde de�i�iklik g�zlenmemi�tir. Ciddi veya orta seviyede b�brek hasar� olan hastalara ait farmakokinetik veriler mevcut de�ildir.
Karaci�er yetmezli�i :
Resmi farmakokinetik �al��ma y�r�t�lmemi�tir. Hafif karaci�er yetmezli�i olan hastalar klinik �al��malara dahil edilmi� ve bu hastalarda OCREVUS'un farmakokineti�inde de�i�iklik g�zlenmemi�tir. Ciddi veya orta seviyede karaci�er hasar� olan hastalara ait farmakokinetik veriler mevcut de�ildir.
5.3. Klinik �ncesi g�venlilik verileri
Klinik d��� veriler; g�venlilik farmakolojisi, tekrarl� doz toksisitesi ve embriyo-f�tal geli�ime y�nelik konvansiyonel �al��malara dayal� olarak insanlar i�in �zel bir risk olmad���n� g�stermektedir. Ocrelizumabla karsinojenisite veya mutajenisite �al��malar� yap�lmam��t�r.
Sinomolgus maymunlar� �zerinde ger�ekle�tirilen iki pre- ve post-natal geli�im �al��malar�nda gestasyonun 20. g�n�nden en az�ndan do�umda kadar ocrelizumab uygulanmas�; glomer�lopati, kemik ili�inde lenfoid folik�l olu�umu, lenfoplazmasitik renal enflamasyon ve yavrunun testis a��rl���nda azalmayla ili�kilendirilmi�tir. Bu �al��malarda uygulanan maternal dozlar, klinik ortamda beklenenden 4,5 ila 21 kat daha y�ksek maksimum ortalama serum konsantrasyonlar�na (C) neden olmu�tur.
�al��mada be� �l�mc�l vaka g�r�lm�� (5/24) olup, biri premat�re do�um nedeniyle g��s�zl�kle birlikte f�rsat�� enfeksiyona, di�eri aktif enfeksiyonlu (mastit) bir anne hayvan�n yavrusunun serebellumunu i�eren bir enfektif meningoensefaliteye ve di�er ��� sar�l�k ve hepatik hasar kan�t�, ��phelenilen viral etiyoloji, muhtemelen bir poliomavir�se atfedilmi�tir. Bu be� do�rulanm�� veya ��phelenilen neonatal enfeksiyonun seyri B h�cre t�kenmesinden potansiyel olarak etkilenmi� olabilir. Ocrelizumab maruz kalm�� anne hayvanlar�n yenido�an yavrular�nda post natal fazda B h�cresi pop�lasyonlar�n�n t�kendi�i fark edilmi�tir. Emzirme d�neminde s�tte �l��lebilir ocrelizumab d�zeyleri saptanm��t�r (serum d�zeylerinde kararl� durumun yakla��k %0,2'si).
6. FARMAS�T�K �ZELL�KLER
6.1. Yard�mc� maddelerin listesi
Sodyum asetat trihidrat (E262)
Glasiyal asetik asit α,α-Trehaloz sihidrat Polisorbat 20 (E432) Enjeksiyonluk su
6.2. Ge�imsizlikler
Bu t�bbi �r�n ile polivinil klor�r (PVC) veya polyolefin (PO) po�etler ve intraven�z uygulamas� setleri aras�nda uyumsuzluk g�zlenmemi�tir.
Bu t�bbi �r�n, B�l�m 6.6'da bahsedilenlerden ba�ka t�bbi �r�nlerle kar��t�r�lmamal�d�r.
6.3. Raf �mr�
A��lmam�� flakon
24 ay
Seyreltilmi� intraven�z inf�zyonluk ��zelti
Kullan�m s�ras�ndaki kimyasal ve fiziksel stabilitesi, 2-8°C'de 24 saat ve oda s�cakl���nda 8 saat s�reyle g�sterilmi�tir.
Mikrobiyolojik a��dan, haz�rlanan inf�zyon hemen kullan�lmal�d�r. Hemen kullan�lmazsa, kullan�m s�ras�ndaki saklama s�releri ve kullan�m �ncesi ko�ullar kullan�c�n�n sorumlulu�undad�r ve seyreltme i�lemi kontrol alt�nda ve valide edilmi� aseptik ko�ullarda yap�lmad��� takdirde, bu s�re normalde 2-8°C'de 24 saati, oda s�cakl���nda 8 saati ge�memelidir.
Bir intraven�z inf�zyonun ayn� g�n tamamlanamamas� durumunda, kalan ��zelti at�lmal�d�r.
6.4. Saklamaya y�nelik �zel tedbirler
Buzdolab�nda saklay�n�z (2ºC – 8 ºC). Dondurmay�n�z.I��ktan korumak i�in flakonlar� karton kutusunda saklay�n�z.
T�bbi �r�n�n seyreltmeden sonraki saklama ko�ullar� i�in, bkz. B�l�m 6.3.
6.5. Ambalaj�n niteli�i ve i�eri�i
Kutuda, florore�ine ile lamine butil kau�uk t�pal�, ALU m�h�rl�, plastik ge�me kapakl�, 10 mL konsantre i�eren 15mL'lik renksiz tip I cam flakon.
1 veya 2 flakonluk ambalajlarda bulunmaktad�r.
6.6. Be�eri t�bbi �r�nden arta kalan maddelerin imhas� ve di�er �zel �nlemler
Seyreltme talimatlar�
�r�n bir sa�l�k uzman� taraf�ndan aseptik teknik kullan�larak haz�rlanmal�d�r. Flakonu �alkalamay�n�z. Seyreltilmi� inf�zyon ��zeltisini haz�rlamak i�in steril bir i�ne ve ��r�nga kullan�lmal�d�r.
�r�n koruyucu madde i�ermez ve sadece tek kullan�m i�indir.
Rengi bozulmu�sa veya yabanc� partik�l madde i�eriyorsa, konsantreyi kullanmay�n�z.
T�bbi �r�n� uygulama �ncesinde seyreltilmelidir. �ntraven�z uygulama i�in ��zeltiler, t�bbi �r�n�n izotonik sodyum klor�r 9 mg/mL (%0,9) enjeksiyon i�in ��zelti (300 mg /250 mL veya 600 mg/500 mL) i�eren bir inf�zyon torbas�nda seyreltilerek yakla��k 1,2 mg/mL ocrelizumab konsantrasyonu elde edilmesiyle haz�rlan�r.
Seyreltilmi� ��zelti, 0,2 veya 0,22 mikron geni�li�inde filtreli bir inf�zyon seti kullan�larak uygulanmal�d�r.
�ntraven�z inf�zyon ba�lat�lmadan �nce, inf�zyon torbas�n�n i�eri�i oda s�cakl���nda olmal�d�r. �mha
Kullan�lmam�� olan �r�nler ya da at�k materyaller, “T�bbi At�klar�n Kontrol� Y�netmeli�i'' ve “Ambalaj ve Ambalaj At�klar�n�n Kontrol� Y�netmeli�i”ne uygun olarak imha edilmelidir.
 Do�um Sonras� Depresyonu
Do�um sonras� depresyonu, do�umdan sonra her on kad�ndan biri taraf�ndan
tecr�be edilen stresli bir durumdur.
Do�um Sonras� Depresyonu
Do�um sonras� depresyonu, do�umdan sonra her on kad�ndan biri taraf�ndan
tecr�be edilen stresli bir durumdur. |
 G�da Alerjisi
Her y�l milyonlarca insan yiyeceklere alerji g�steriyor.
G�da Alerjisi
Her y�l milyonlarca insan yiyeceklere alerji g�steriyor. |
 |
Parkinson Hastal��� Hastal�k ilk kez 1817 de �ngiliz doktor James Parkinson taraf�ndan tan�mlanm�� ve Dr. Parkinson hastal��� “sallay�c� fel�” olarak kaleme alm��. |
 |
Grip, So�uk Alg�nl��� ve �ks�r�k Grip ve so�uk alg�nl��� (nezle) semptomlar� aras�ndaki fark� bilmek �nemlidir. So�uk alg�nl��� gripten daha hafif belirtiler g�steren bir solunum yolu hastal���d�r. |
 |
S�rt A�r�s� S�rt a�r�s� birden bire ortaya ��k�p �iddetli (akut) olabilir veya zamanla geli�ip daha uzun s�reli sorunlara (kronik) neden olabilir. |
�LA� GENEL B�LG�LER�
Roche M�stahzarlar� Sanayi A.�.
| Geri �deme Kodu | A16804 |
| Sat�� Fiyat� | 96031.28 TL [ 15 Apr 2024 ] |
| �nceki Sat�� Fiyat� | 96031.28 TL [ 5 Apr 2024 ] |
| Original / Jenerik | Original �la� |
| Re�ete Durumu | Normal Re�eteli bir ila�d�r. |
| Barkodu | 8699505761978 |
| Etkin Madde | Okrelizumab |
| ATC Kodu | L04AA36 |
| Birim Miktar | 300 |
| Birim Cinsi | MG |
| Ambalaj Miktar� | 1 |
| Antineoplastik ve �mm�nomod�lat�r Ajanlar > �mm�nsupresif Ajanlar |
| �thal ( ref. �lke : Isvicre ) ve Be�eri bir ila�d�r. |